
Aortic coarctation

For patient information click here
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Synonyms and keywords: coarct; coarctation of the aorta
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Overview
Aortic coarctation is a localized narrowing or abrupt constriction of the aortic arch anywhere along its length. It is most common distal to the origin of the left subclavian artery, near the area where the ductus arteriosus (ligamentum arteriosum after its regression) inserts. Less commonly, the obstruction can occur in the abdominal aorta. Coarctation may be associated with a bicuspid aortic valve. There is a dilation of the aorta immediately above the narrowing, but especially just below. Therefore the latin term coarctatus, which means contracted or tightened.
Historical Perspective
Aortic coarctation has been on historical record since as early as 1760. In the 1900s, researchers such as Bonnett and Johnson would later further classify the anatomical configurations associated with aortic coarctation.
Classification
An aortic coarctation can be classified in three ways depending on the anatomical configuration. These include: preductal coarctation, ductal coarctation, and postductal coarctation. All classifications involve narrowings of the aorta that directly impact the aortic hemodynamics.
Pathophysiology
An aortic coarctation results from both, congenital and acquired means. Factors directly influencing the pathophysiology include defect location and sites of secondary dilation.
Causes
Like many congenital heart diseases, the cause of aortic coarctation is not clear. The etiology of coarctation of the aorta may be explained by multifactorial inheritance hypothesis. Clinical studies suggest that genetic, familial influence and environmental factors both play an important role during pregnancy. It has been found to be associated more with patients with Turner syndrome. Additional research suggests a possible link between other congenital heart diseases and an aortic coarctation, indicating that those with congenital heart disease are more likely to have an accompanying secondary defect.
Differentiating Aortic Coarctation from other Diseases
A thorough examination is necessary to truly diagnose an aortic coarctation. Conditions with similar symptoms to an aortic coarctation include: aortic stenosis, cardiomyopathies (dilated cardiomyopathy and hypertrophic cardiomyopathy), endocardial fibroelastosis, primary hypertension, hypoplastic left heart syndrome, viral myocarditis, congenital adrenal hyperplasia, patent ductus arteriosus, polyarteritis, sepsis, and shock from a variety of causes.
Epidemiology and Demographics
Coarctation of the aorta is a common malformation. It occurs in about 7% of patients with congenital heart defects. It is more common in males than females with a ratio of 2:1. Up to 25% of patients with Turner syndrome have coarctation of the aorta. It is more common in caucasians with approximately 7 times more cases in caucasians versus asian.
Risk Factors
Although the cause of aortic coarctation is not definitively known, certain factors have been associated with a potential risk increase. These include genetic anomalies such as Turner Syndrome, familial history, environmental factors (viral infections during pregnancy), and neonatal care.
Screening
Arterial hypertension in the right arm with normal to low blood pressure in the lower extremities should prompt consideration of the diagnosis of aortic coarctation.
Natural History, Complications and Prognosis
An aortic coarctation is generally symptomatic enough during childhood to prompt evaluation, and approximately 80% of cases are diagnosed during childhood. The remaining 20% of cases are often less symptomatic with less severe narrowing, but will ultimately require correction or irreversible organ damage can occur. Common complications, when left untreated, include: aortic rupture, infective endocarditis, congestive heart failure, and calcification of the aorta.
Diagnosis
History and Symptoms
Common symptoms include dizziness, fainting (or frank syncope), shortness of breath (dyspnea), severe headache most likely due to hypertension, chest pain (angina), cold feet/legs, nosebleed, leg cramps with exercise, high blood pressure (hypertension) with exercise, decreased ability to exercise, and poor growth.
Physical Examination
Physical examination acts as an important tool in the diagnosis of coarctation of aorta. Differential hypertension (depending on the location of coarctation) is often present with increased blood pressure in the upper extremities and relative hypotension in the lower extremities. The difference is usually in systolic blood pressure whereas the diastolic blood pressures are typically similar. Similarly, the pulses in upper extremities are bounding whereas the femoral pulses are often diminished (brachial-femoral delay]). There are 3 potential sources of a murmur: multiple arterial collaterals (continuous murmur), an associated bicuspid aortic valve (systolic ejection click), and the coarctation itself which can be heard over the left infraclavicular area and under scapula. Murmurs due to associated cardiac abnormalities such as VSD or aortic valve stenosis, may also be detected. Neonates may present with discrepancies in blood pressure and pulses between the limbs, differential cyanosis or reversed differential cyanosis (depending on associated lesions), murmur, congestive heart failure, and shock. Older children and adolescent may be referred due to agitated behavior, headache, vision problems, and hypertension.
Electrocardiogram
Electrocardiogram may be used as a diagnostic tool in the evaluation of an aortic coarctations. ECG findings associated with an aortic coarctation depend on the severity of the coarctation. Milder cases may show signs of a normal ECG. However, more severe coarctations will have abnormal ECGs showing evidence of left ventricular hypertrophy.
Chest X Ray
Aortic coarctation on chest X ray presents with irregular notching of the inferior margins of the posterior ribs resulting from collateral flow through dilated and pulsatile intercostal arteries. An inverted “3” sign of the barium-filled esophagus or a “3” sign on a highly penetrated chest radiograph may be visualized. There are often signs of congestive heart failure including cardiomegaly, pulmonary edema, and prominent pulmonary vasculature on the chest X ray.
MRI
Magnetic resonance imaging (MRI) can define the location and severity of a coarctation. MRI can also detect associated cardiac abnormalities and is used for serial follow-up after surgical repair or balloon angioplasty. MRI is recommended to look for aneurysm formation following repair of a coarctation. MR angiography has almost completely replaced invasive catheter based techniques for evaluating re coarctation. In adults with untreated coarctation blood often reaches the lower body through collaterals, eg. internal thoracic arteries via the subclavian arteries. These can be visualized on MR or angiography.
CT
CT is useful in older or postoperative patients as it helps in to assessing residual obstruction, hypoplasia, aneurysms, multiple surgical clips or a stent present in the area of coarctation. [1]
Echocardiography
Echocardiography is an useful diagnostic tool for coarctation of aorta. The 2 D echocardiography helps in the evaluation of the aortic arch to assess the transverse aortic arch, isthmus, severity of coarctation, and other associated cardiac abnormalities. Doppler echocardiography determines the gradient across the coarctation and guides decisions regarding treatment.
Treatment
Medical Therapy
The treatment choice depends on the patients age of presentation, severity, the location of the coarctation and other associated anomalies. For children who present early, the role of medical management lies in stablizing the patient for surgery. However, in older kids and adolescent presenting with hypertension treatment is guided towards correction of hypertension and other associated anomalies. Beta blocker is treatment of choice for both pre and post operative hypertension.
Surgery
Therapy/Treatment is conservative if asymptomatic, but may require surgical resection of the narrow segment if there is arterial hypertension. The first operations to treat coarctation were carried out by Clarence Crafoord in Sweden in 1944.[2] In some cases angioplasty can be performed to dilate the narrowed artery. If the coarctation is left untreated, arterial hypertension may become permanent due to irreversible changes in some organs (such as the kidney).
Videos
{{#ev:youtube|o_mWpuR3Z-Y}}
References
- ↑ Mohiaddin RH, Kilner PJ, Rees S, Longmore DB (1993). “Magnetic resonance volume flow and jet velocity mapping in aortic coarctation”. Journal of the American College of Cardiology. 22 (5): 1515–21. PMID 8227813. Retrieved 2012-04-14. Unknown parameter
|month=ignored (help) - ↑ Radegran, Kjell (2003), “The early history of cardiac surgery in Stockholm”, Journal of Cardiac Surgery, 18 (6): 564–572, doi:10.1046/j.0886-0440.2003.02071.x, ISSN 0886-0440, PMID 14992112, retrieved 2009-03-10
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Overview
Aortic coarctation was first described as early as 1760. In the 1900s Bonnett and Johnson further classified the anatomical variants associated with aortic coarctation.
Historical Perspective
1760
- Johann Friederich Meckel, Prussian anatomist, introduced the description of coarctation of aorta in 1760.
1903
- In 1903, Bonnett was the first in classify the aortic coarctation into infantile type (diffuse narrowing of the aorta in the region proximal to the ductus arteriosus) and adult type or more abrupt constriction of the aorta just distal to the origin of the left subclavian artery, close to the ductus insertion.
1951
- In 1951, Johnson et al, classified the coarctation based upon the position of the ductus arteriosus and its relation to the coarctation as preductal aortic coarctation and postductal aortic coarctation.
References
Classification
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Overview
Aortic coarctation can be classified as preductal coarctation, ductal coarctation, and postductal coarctation depending upon the coarctation’s anatomic relationship to the ductus arteriosus. All classifications involve narrowings of the aorta that directly impact the aortic hemodynamics.
Classification

Schematic drawing of alternative locations of a coarctation of the aorta, relative to the ductus arteriosus. A: Ductal coarctation, B: Preductal coarctation, C: Postductal coarctation. 1: Aorta ascendens, 2: Arteria pulmonalis, 3: Ductus arteriosus, 4: Aorta descendens, 5: Truncus brachiocephalicus, 6: Arteria carotis communis sinistra, 7: Arteria subclavia sinistra[1]
There are three variations of coarctation of the aorta:
Preductal Coarctation
The narrowing is proximal to the ductus arteriosus. If severe, blood flow to the aorta distal (to lower body) to the narrowing is dependent on a patent ductus arteriosus, and hence its closure can be life-threatening. Preductal coarctation results when an intracardiac anomaly during fetal life decreases blood flow through the left side of the heart, leading to hypoplastic development of the aorta.
Ductal Coarctation
The narrowing occurs at the insertion of the ductus arteriosus. This kind usually appears when the ductus arteriosus closes.
Postductal Coarctation
The narrowing is distal to the insertion of the ductus arteriosus. Even with an open ductus arteriosus blood flow to the lower body can be impaired. Newborns with this type of coarctation may be critically sick from birth. This type is most common in adults. It is associated with notching of the ribs, hypertension in the upper extremities, and weak pulses in the lower extremities. Postductal coarctation is most likely the result of muscular ductal (ductus arteriosis) extension into the aorta during fetal life.
References
- ↑ Valdes-Cruz LM, Cayre RO: Echocardiographic diagnosis of congenital heart disease. Philadelphia, 1998.
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Overview
An aortic coarctation results from both, congenital and acquired means. Factors directly influencing the pathophysiology include defect location and sites of secondary dilation.
Pathophysiology
-
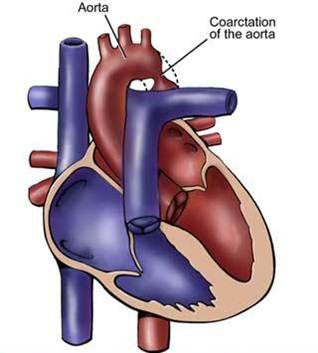 Coarctation of the descending aorta.
Coarctation of the descending aorta. -
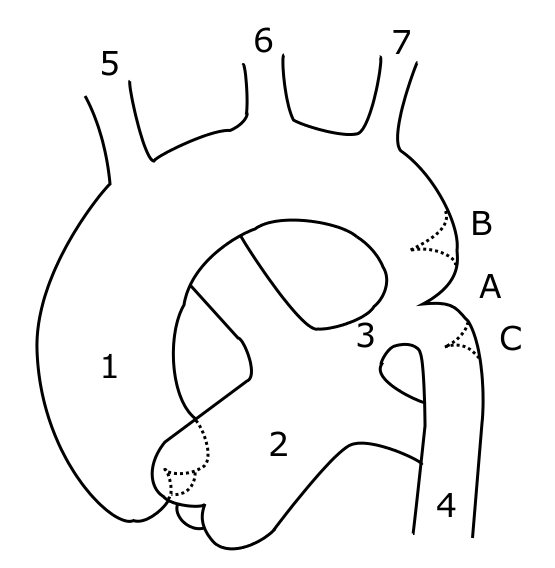 Schematic drawing of alternative locations of a coarctation of the aorta, relative to the ductus arteriosus. A: ductal coarctation, B: preductal coarctation, C: postductal coarctation. 1: Aorta ascendens, 2: Arteria pulmonalis, 3: Ductus arteriosus, 4: Aorta descendens, 5: Trunchus brachiocephalicus, 6: Arteria carotis communis sinister, 7: Arteria subclavia sinister
Schematic drawing of alternative locations of a coarctation of the aorta, relative to the ductus arteriosus. A: ductal coarctation, B: preductal coarctation, C: postductal coarctation. 1: Aorta ascendens, 2: Arteria pulmonalis, 3: Ductus arteriosus, 4: Aorta descendens, 5: Trunchus brachiocephalicus, 6: Arteria carotis communis sinister, 7: Arteria subclavia sinister


Coarctation of the aorta can be:
- Congenital coarctation resulting from an infolding of the aortic media that incorportaes ductal tissue, forming a ridge that eccentrically narrows the lumen of the vessel. Subsequent intimal proliferation on the ridge leads to progressive narrowing of the vessel lumen. There is a dilatation before and after the narrowing, giving the aorta an hourglass appearance. The exact etiology of the aortic abnormality remains unclear but likely involves a defect in the vascular wall of the aorta due to reduced antegrade intrauterine blood flow or to constriction of ductal tissue extending into the thoracic aorta.
- Acquired coarctation occurring in systemic arteritides such as Takayasu arteritis. Additionally it may occur in rare cases of severe atherosclerosis.
Defect Location

- 95% of the lesions are located distal to the left subclavian artery and proximal to the ductus arteriosus (preductal coarctation) or just at or distal to the ductus (postductal coarctation).
- 5% of coarctations are located proximal to the left subclavian artery, or rarely in the abdominal aorta.
- In some cases, coarctation presents as a long segment or a tubular hypoplasia.
- The stenosis is caused by an infolding of the left posterolateral aspect of the aortic wall resulting in an eccentric narrowing.
Sites of Secondary Dilation
- Aorta proximal to the coarct
- Aorta distal to the coarctation
- Left subclavian artery
- The narrowing progresses throughout life, and extensive collaterals develop from the subclavian (predominantly) and axillary arteries through:
- Internal mammary artery
- Scapular artery
- Intercostal arteries
- Epigastric arteries
- Anterior spinal arteries
Genetics
- Aortic coarctation, like many congenital heart diseases, is more common in patients with other genetic conditions.
- As many as 10-25% of patients with Turner syndrome have an accompanying coarctation of the aorta.
Gross Pathology
-
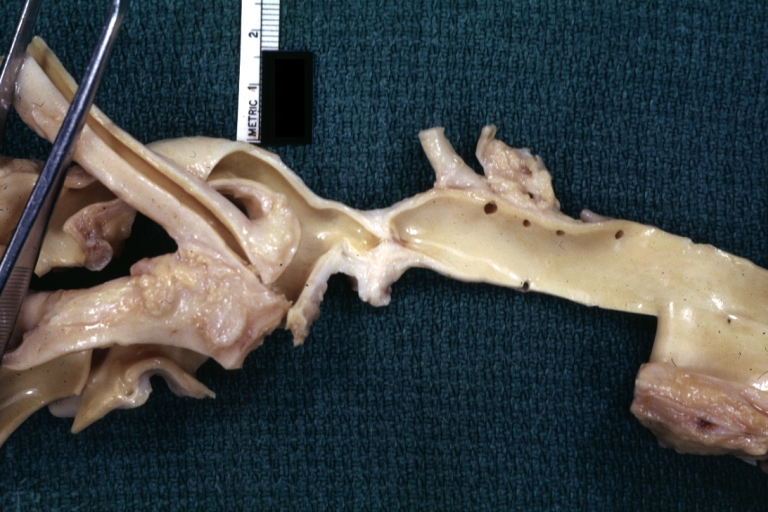 AORTA: Coarctation, Adult: Gross, fixed tissue, an excellent illustration of postductal coarctation
AORTA: Coarctation, Adult: Gross, fixed tissue, an excellent illustration of postductal coarctation -
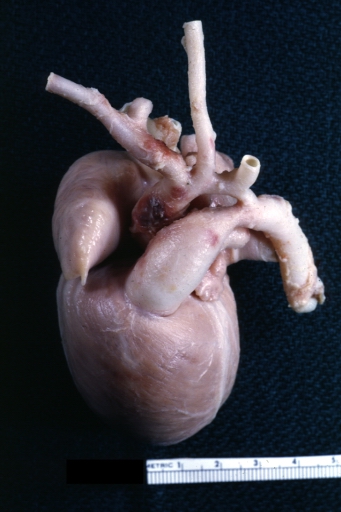 AORTA: Coarctation: Gross, hypoplastic aortic arch and infantile coarctation well demonstrated.
AORTA: Coarctation: Gross, hypoplastic aortic arch and infantile coarctation well demonstrated. -
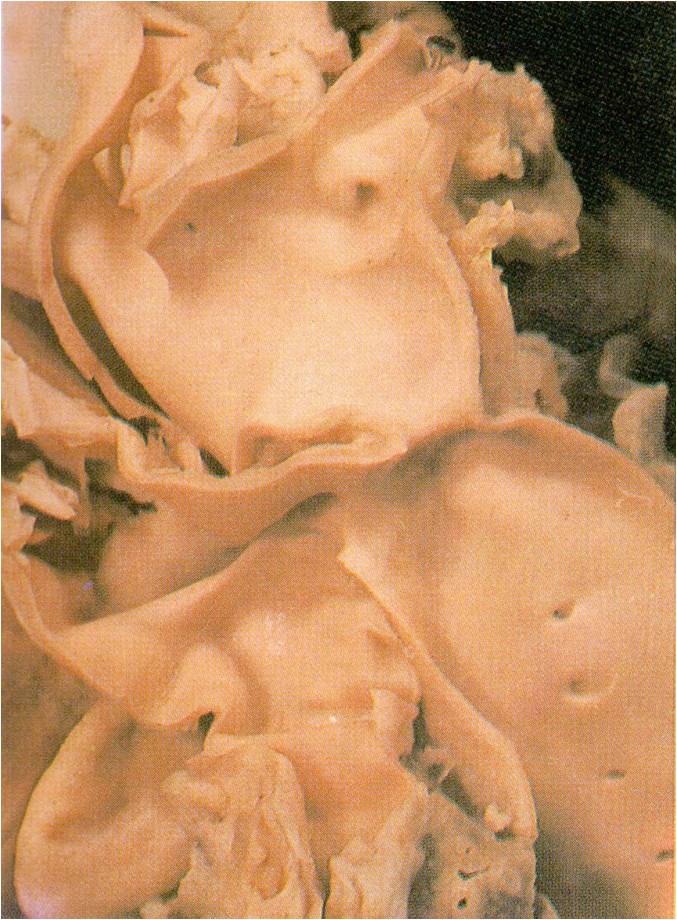 Localized Coarctation of the aorta.
Localized Coarctation of the aorta.



Associated Conditions
- It is commonly associated with bicuspid aortic valve.
- There is 5 fold increase in the intracranial aneurysm in patient with coarctation.
Videos
{{#ev:youtube|SiNJfvK_qeI}}
References
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Overview
Like many congenital heart diseases, the cause of aortic coarctation is not clear. The etiology of coarctation of the aorta may be explained by multifactorial inheritance hypothesis. Clinical studies suggest that genetic, familial influence and environmental factors both play an important role during pregnancy. It has been found to be associated more with patients with Turner syndrome. Additional research suggests a possible link between other congenital heart diseases and an aortic coarctation, indicating that those with congenital heart disease are more likely to have an accompanying secondary defect.
Causes
- The etiology of coarctation of the aorta may be explained by multifactorial inheritance hypothesis.
- Aortic coarctation is more common in persons with certain genetic disorders, such as Turner syndrome.
- It can also be due to birth defects of the aortic valves. Aortic coarctation is one of the more common heart conditions that are present at birth (congenital heart conditions). It is usually diagnosed in children or adults under age 40. Coarctation of the aorta may be seen with other congenital heart defects, such as: bicuspid aortic valve, defects in which only one ventricle is present, ventricular septal defect
- It has been found to have increased incidences in some families.
- Maternal drug use – Clomifene
References
Differentiating Aortic Coarctation from other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Overview
A thorough examination is necessary to truly diagnose an aortic coarctation. Conditions with similar symptoms to an aortic coarctation include: aortic stenosis, cardiomyopathies (dilated cardiomyopathy and hypertrophic cardiomyopathy), endocardial fibroelastosis, primary hypertension, hypoplastic left heart syndrome, viral myocarditis, congenital adrenal hyperplasia, patent ductus arteriosus, polyarteritis, sepsis and shock
Differentiating Aortic Coarctation from other Diseases
A number of conditions are associated with the aortic coarctation. During an examination, it is important to be cognizant that the following conditions need to be differentiated from an aortic coarctation:
- Aortic stenosis
- Cardiomyopathies (dilated cardiomyopathy and hypertrophic cardiomyopathy)
- Endocardial fibroelastosis
- Essential hypertension
- Hypoplastic left heart syndrome
- Viral myocarditis
- Circulatory collapse due to hypoadrenalism, congenital adrenal hyperplasia
- Patent ductus arteriosus
- Hypertension
- Polyarteritis
- Sepsis
- Shock
References
Epidemiology and Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Cafer Zorkun, M.D., Ph.D. [2], Priyamvada Singh, M.B.B.S.[3]
Overview
Coarctation of the aorta is a common congenital malformation. It occurs in about 7% of patients with congenital heart defects. It is more common in males than females with a ratio of 2:1. Up to 25% of patients with Turner syndrome have coarctation of the aorta. It is 7 times more common among caucasians than asians.
Epidemiology and Demographics
Age
- The diagnosis is often missed in first year of life.
- Generally, patients with coarctation of the aorta present early in life with congestive heart failure or later in life with hypertension.
Gender
- It is 2 times more common in males than females.
Race
- Coarctation is 7 times more common among Caucasians than Asians.
- The incidence is lower in Native Americans than Caucasians.
United States of America
- Aortic coarctation is a common heart defect.
- It forms approximately 6-10% of all congenital heart disease cases.
- In live births, it accounts for approximately 5-7% of congenital heart disease in severely ill infants.
International
- The prevalence is lower in Asian countries compared to American and European countries.
References
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Overview
Risk factors associated with an increased risk of coarctation include genetic anomalies, familial history, environmental factors, and neonatal care.
Risk Factors
Like many congenital heart disease, the cause of aortic coarctation is not well defined. Clinical studies suggest that genetic and environmental factors both play an important role during pregnancy. These include:
- Genetic disorders, such as Turner syndrome. As many as 10-25% of patients with Turner syndrome have an accompanying coarctation of the aorta.
- Gender, more common in males than females (2:1 ratio).
- Viral infections during pregnancy.
- Presence of another congenital heart disease, such as:
References
Screening
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1], Associate Editor-in-Chief: Cafer Zorkun, M.D., Ph.D. [2]
Overview
Arterial hypertension in the right arm with normal to low blood pressure in the lower extremities should prompt consideration of the diagnosis of aortic coarctation.
Screening
Following findings may be observed during screening of aortic coarctation;
- Blood pressure: Arterial hypertension in the right arm with normal to low blood pressure in the lower extremities is classic. The blood pressure is higher in the upper extremities than in the lower extremities. The patient may complain of a headache due to hypertension.
- Pulses: Femoral pulses are often diminished in strength. Exercise exacerbates this gradient. If the coarctation is situated before the left subclavian artery, the left pulse will be diminished in strength and asynchronous radial pulses will be detected in the right and left arms. A radial-femoral delay between the right arm and the femoral artery may be apparent, while no such delay may be observed with left arm radial-femoral palpation. A coarctation occurring after the left subclavian artery will produce synchronous radial pulses, but radial-femoral delay will be present under palpation in either arm.
- Neck: There may be webbing of the neck in patients with Turner syndrome, 10% of whom have aortic coarctation.
- Heart
- A systolic ejection click is present when there is an associated bicuspid aortic valve.
- The S2 is loud secondary to hypertension.
- An S4 may be present secondary to LVH.
- There are 3 potential sources of a murmur: arterial collaterals, an associated bicuspid aortic valve, and the coarctation itself which can be heard over the spine.
- A prominent P2 may be present if there is associated pulmonary hypertension.
- Extremities
- Cyanosis of the lower extremities may be present.
- Occasionally adults may have narrow hips and thin legs or have an undeveloped left arm (in those patients in which the coarctation compromises the origin of the subclavian artery).
References
Natural History, Complications and Prognosis
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Priyamvada Singh, M.B.B.S.[2], Cafer Zorkun, M.D., Ph.D. [3]; Assistant Editor(s)-In-Chief: Kristin Feeney, B.S.[4]
Overview
80% of patients are diagnosed during childhood. In the remaining 20% of cases, the symptoms are often less severe, but the coarctation will ultimately require correction in order to prevent irreversible organ damage. Common complications among patients who go untreated include: aortic rupture, infective endocarditis, congestive heart failure, and calcification of the aorta.
Natural History
In infants with a preductal coarct, the LV output goes to the upper extremities, and the RV output goes to the lower extremities through the patent ductus.
- Childhood:
- 80% of cases are diagnosed in childhood.
- The preductal form is usually discovered in early infancy because it is usually severe.
- Childhood coarctation is associated with a ventricular septal defect (VSD), tubular hypoplasia of the aortic arch, transposition of the great vessels, and mitral valve disease.
- Adolescence:
- When first recognized in adolescents, coarctation of the aorta is generally asymptomatic.
- Adulthood:
- The postductal form is often less severe and discovered in adulthood.
- Infrequently coarctation of the aorta is associated with other congenital abnormalities.
- In patients over the age of 30, major complications leading to death are not uncommon. [1][2]
- 75% of patients with coarctation will have hypertension at 30 years of their age.
- If the coarctation is left untreated, arterial hypertension may become permanent due to irreversible changes in some organs (such as the kidney).
Complications
About 50% of patients with coarctation of the aorta die within the first three decades of life, and more than 75% are dead by age 50 due to:
- Rupture of the aorta or aortic dissection
- Most frequently in the third or fourth decade.
- Dissections originate either proximally (secondary to hypertension and local stress)or distally (where the jet erodes the intima).
- Ruptures may bleed into the esophagus, and hematemesis or melena may portend disaster.
- Infective endocarditis or endarteritis
- Most frequently in the second to fourth decade of life.
- Rupture of the circle of Willis
- Most frequently in the second or third decade of life.
- Secondary to the increased incidence of aneurysms in this population and the presence of proximal hypertension.
- Congestive heart failure
- Common in infants, often occurs with associated abnormalities such as VSD or mitral valve disease.
- In the adult is secondary to hypertension associated with coronary artery disease or aortic valve disease.
- Calcified aortic stenosis
- Result of associated bicuspid aortic valve disease that over time becomes calcified.
- Premature death occurs due to
Prognosis
Prognosis is variable. The prognosis of aortic coarctation depends on whether balloon angioplasty and stenting or the surgery has been done or not.
- The mortality rate of aortic coarctation depends largely on the age of surgical repair. Left untreated, less than 20% of untreated patients live to the age of 50. However, with surgical repair, mortality rates are far lower and survival is prolonged.
- If repaired by the age of 14, the mortality rate during the 20 years following is only 9% (a 91% survival rate).
- If repaired later than age 14, the mortality rate is 11% (a 79% survival rate).
- Among expectant mothers with a coarctation of the aorta, the maternal mortality rate ranges between 3-8%. Even with repair, there is a risk of developing complications during pregnancy.
References
Diagnosis
Diagnosis
History and Symptoms | Physical Examination | Electrocardiogram | Chest X Ray | CT | MRI | Angiography | Echocardiography
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH