
Wilson's disease
For patient information click here
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Cafer Zorkun, M.D., Ph.D. [2], Ahmed Elsaiey, MBBCH [3]
Synonyms and keywords: Hepatolenticular degeneration
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Wilson’s disease is a rare autosomal recessive inherited disease that occurs in both children and adults due to copper accumulation in different organs, mainly in the liver and the brain. Wilson’s disease is caused by genetic mutations in the ATP7B gene which is responsible for the transportation of the dietary copper to the liver. Copper is normaly absorbed in the stomach and transported to the liver in order to function properly as a cofactor for some enzymes as the cytochrome c oxidase enzyme. Copper is also incorporated in the formation of ceruloplasmin in the Trans Golgi network. Transportation of the copper to the Trans Golgi network occurs via ATOX1 protein and others are bound to metallothionein. Pathogenesis of Wilson’s disease include any impairment that may occur during copper transportation and incorporation into different essential metabolic mechanisms. The mechanisms responsible for the pathogenesis of Wilson’s disease include high serum copper level, accumulation of the copper in the liver, and deposition of the copper in different organs resulting in ultimate injury and organ damage. The prevalence of Wilson’s disease is estimated to be 3 cases per 100,000 individuals worldwide. Wilson’s disease affects individuals between 5 to 35 years old. Common risk factors of Wilson’s disease include newborns of disease-carrier parents. Less common risk factors include excess non-vegetarian diet. If left untreated, Wilson’s disease will lead to death as the copper accumulation in the liver and brain results in cirrhosis and severe dystonia, respectively. Common complications of Wilson’s disease include hepatocellular carcinoma, renal failure, and persistent neurological manifestations. Prognosis of Wilson’s disease is usually good in case of early detection and proper treatment. Patients with Wilson’s disease may remain asymptomatic until the copper deposits in the liver and brain mainly. Common hepatic symptoms include abdominal distension, abdominal pain, fatigue, bleeding tendency, and esophageal varices. Common neuropsychiatric symptoms include tremors, ataxia, dysarthria, and impulsiveness. Physical examination of patients with Wilson’s disease is usually remarkable for jaundice and easy bruising in the skin. Physical examination also is remarkable for Kayser-Fleischer ring in the eyes as a result of the copper accumulation in the cornea. Laboratory findings suggestive for Wilson’s disease include low ceruloplasmin level, high serum copper concentration and high urinary excretion of the copper. MRI scanning can be used in detecting brain abnormalities especially in the basal ganglia. Medical therapy is the mainstay of therapy for treatment patients with Wilson’s disease. The treatment option include penicillamine, trientine, and Zinc. Liver transplantation is reserved for the patients that present with acute liver failure and unresponsive to the medical treatment.
Historical Perspective
Wilson’s disease was first described by Dr. Samuel Alexander Kinnier Wilson at which he described the pathological changes in the brain and liver in 1912. Many research studies revealed the correlation between ATP7B gene mutation and Wilson’s disease. The first effective oral chelator was discovered by Dr. Walshe in 1956.
Classification
Wilson’s disease is classified based on the symptomatic presentation into hepatic and neurologic Wilson’s disease.
Pathophysiology
Copper is one of the essential elements for the human body. Copper must be absorbed and transported in order to function properly. Copper is transported bound to metallothionein or carried by ATOX1 to the trans-Golgi network. Impairment of copper incorportation in formation of ceruloplasmin will lead to increase the serum level of the copper. The increase of copper level will lead to accumulation of the excess amount in the liver and other organs. ATP7B gene mutation is found in the majority of the patients with Wilson’s disease.
Causes
Wilson’s disease is caused by ATP7B gene mutation and impairement of copper transportation.
Differentiating Wilson’s disease from Other Diseases
Wilson’s disease must be differentiated from other diseases that cause jaundice like hemochromatosis, viral hepatitis, alcoholic hepatitis, drug induced hepatitis, and autoimmune hepatitis.
Epidemiology and Demographics
The prevalence of Wilson’s disease is estimated to be 3 cases per 100,000 individuals worldwide. Wilson’s disease affects individuals between 5 to 35 years old.
Risk Factors
Commpn risk factors of Wilson’s disease include newborns of disease-carrier parents. Less common risk factors inlcude excess non-vegetarian diet.
Screening
There is insufficient evidence to recommend routine screening for Wilson’s disease.
Natural History, Complications, and Prognosis
If left untreated, Wilson’s disease will lead to death as the copper accumulation in the liver and brain results in cirrhosis and severe dystonia respectively. Common complications of Wilson’s disease include hepatocellular carcinoma, renal failure, and persistent neurological manifestations. Prognosis of Wilson’s disease is usually good in case of early detection and proper treatment.
Diagnosis
Diagnostic study of choice
Wilson’s disease diagnostic criteria is based on scoring system which includes clinical presentation, laboratory findings, and gene mutation analysis. The score of 4 or more is diagnostic for Wilson’s disease. The score of 2 or less is ruling out Wilson’s disease.
History and Symptoms
Patients with Wilson’s disease may remain asymptomatic until the copper deposits in the liver and brain mainly. Common hepatic symptoms include abdominal distension, abdominal pain, fatigue, bleeding tendency, and esophageal varices. Common neuropsychiatric symptoms include tremors, ataxia, dysarthria, and impulsiveness. Less common symptoms of wilson’s disease include urolithiasis and hematuria.
Physical Examination
Patients with Wilson’s disease usually appear tired. Physical examination of patients with Wilson’s disease is usually remarkable for jaundice and easy bruising in the skin. Physical examination also is remarkable for Kayser-Fleischer ring in the eyes as a result of the copper accumulation in the cornea. Common physical examination findings in the abdomen include abdominal tenderness, ascites, and spider angiomata. Common neuropsychiatric signs include seizures, parkinsonism like signs, depression, and anxiety.
Laboratory Findings
Laboratory findings suggestive for Wilson’s disease include low ceruloplasmin level, high serum copper concentration, and high urinary excretion of thecopper.
Electrocardiogram
There are no EKG findings associated with Wilson’s disease. However, if the heart is affected by Wilson’s disease, EKG should be performed to exclude any concurrentarrhythmias.
X-ray
There are no x-ray findings associated with Wilson’s disease.
Ultrasound
There are no ultrasound findings associated with Wilson’s disease.
CT scan
CT scan can be used in detecting abnormalities in the brain but MRI is more sensetive in diagnosing Wilson’s disease associated with neurological manifestations.
MRI
There are no specific MRI findings associated with Wilson’s disease especially in cases who present with only hepatic manifestations. However, it may show abnormalities in the basal ganglia in the patients who presented with neuropsychiatric manifestations.
Other Imaging Findings
There are no other imaging findings associated with Wilson’s disease.
Other Diagnostic Studies
Liver biopsy is performed in suspected cases of Wilson’s disease as it may show many histopathological features. Liver biopsy may show mild steatosis, hepatocellular necrosis, macronodular cirrhosis, and fulminant liver failure features as parenchymal collapse. Genetic testing is also recommended in Wilson’s disease to obtain the family history of the disease and for early detection.
Treatment
Medical Therapy
Medical therapy is the mainstay of therapy for treatment patients with Wilson’s disease. The treatment option include copper chelators as penicillamine and trientine. The treatment also includes Zinc to prevent copper reaccumulation.
Surgery
Surgery is not the first-line treatment option for patients with Wilson’s disease. Surgery is usually reserved for patients with acute hepatitis and unresponsive to the medical therapy. Liver transplantation is recommended in patients with acute liver failure.
Primary prevention
Effective measures for the primary prevention of Wilson’s disease include genetic analysis of the family members in order to early detect and treat the disease.
Secondary prevention
The primary and secondary prevention strategies for Wilson’s disease are the same.
References
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Wilson’s disease was first described by Dr. Samuel Alexander Kinnier Wilson where he described the pathological changes in the brain and liver in 1912. Many research studies revealed the correlation between ATP7B gene mutation and Wilson’s disease. The first effective oral chelator was discovered by Dr. Walshe in 1956.
Historical perspective
Discovery
- In 1912, the neurologist Dr. Samuel Alexander Kinnier Wilson was the first to describe Wilson’s disease. Dr. Wilson described the pathological changes in the brain and liver.[1]
- In 1902, Dr. Kayser described the corneal rings which was abnormal pigmentation of the eye which was named after that Kayser-Fleisher rings. Dr. Wilson did not correlate between the disease and the pigmented corneal rings at the time he discovered the disease.[2]
- In 1948, Dr. John N. Cumings described the correlation between the copper accumulation and the pathological changes that occur in the liver and the brain.[3]
- From 1980s to 1990s, many research studies proved the linkage between the mutation of ATP7B gene and Wilson’s disease.[4][5]
Landmark Events in the Development of Treatment Strategies
- In 1951, Dr. Cumings and Dr. Denny-Brown reported the first effective treatment against Wilson’s disease by the copper chelators.[6]
- In 1956, Dr. Walshe discovered penicillamine which was the first effective oral chelator back then.[7]
- In 1961, Dr. Schouwink and Dr. Hoogenraad used Zinc acetate as a medical therapy in treatment of Wilson’s disease.
- In 1982, Dr. Walshe also described trientine as an effective chelator also for the copper.[8]
References
- ↑ Kinnier Wilson SA (1912). “Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver” (PDF). Brain. 34 (1): 295–507. doi:10.1093/brain/34.4.295.
- ↑ Lorincz MT (2010). “Neurologic Wilson’s disease”. Ann N Y Acad Sci. 1184: 173–87. doi:10.1111/j.1749-6632.2009.05109.x. PMID 20146697.
- ↑ Cumings JN (1948). “The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration” (PDF). Brain. 71 (Dec): 410–5. doi:10.1093/brain/71.4.410. PMID 18124738.
- ↑ Tanzi RE, Petrukhin K, Chernov I; et al. (1993). “The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene”. Nat. Genet. 5 (4): 344–50. doi:10.1038/ng1293-344. PMID 8298641.
- ↑ Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993). “The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene”. Nat. Genet. 5 (4): 327–37. doi:10.1038/ng1293-327. PMID 8298639.
- ↑ Cumings JN (1951). “The effects of B.A.L. in hepatolenticular degeneration”. Brain. 74 (1): 10–22. doi:10.1093/brain/74.1.10. PMID 14830662. Unknown parameter
|month=ignored (help) - ↑ Walshe JM (1956). “Wilson’s disease; new oral therapy”. Lancet. 267 (6906): 25–6. doi:10.1016/S0140-6736(56)91859-1. PMID 13279157. Unknown parameter
|month=ignored (help) - ↑ Walshe JM (1982). “Treatment of Wilson’s disease with trientine (triethylene tetramine) dihydrochloride”. Lancet. 1 (8273): 643–7. doi:10.1016/S0140-6736(82)92201-2. PMID 6121964. Unknown parameter
|month=ignored (help)
Classification
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Wilson’s disease is classified based on the symptomatic presentation into hepatic and neurologic Wilson’s disease.
Classification
Wilson’s disease is classified based on the symptomatic presentation as the following:[1]
- Hepatic Wilson’s disease (H):
- H1: Acute hepatic Wilson’s disease
- H2: Chronic hepatic Wilson’s disease
- Neurologic Wilson’s disease (N):
- N1: Neurologic manifestations associated with hepatic involvement
- N2: Neurologic manifestations only
- NX: Unknown liver involvement
References
- ↑ Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I; et al. (2003). “Diagnosis and phenotypic classification of Wilson disease”. Liver Int. 23 (3): 139–42. PMID 12955875.
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Copper is one of the essential elements for the human body. Copper must be absorbed and transported in order to function properly. Copper is transported bound to metallothionein or carried by ATOX1 to the trans-Golgi network. Impairment of copper incorportation in formation of ceruloplasmin will lead to increase the serum level of the copper. The increase of copper level will lead to accumulation of the excess amount in the liver and other organs. ATP7B gene mutation is found in the majority of the patients with Wilson’s disease.
Pathophysiology
Normal copper transportation and metabolism
- Copper is one of the essential elements that is required daily in diet in a range of 1.5 to 2 mg.[1]
- Copper is incorporated in number of important functions in the body. It functions as a component of some enzymes as cytochrome c oxidase, dopamine β-hydroxylase, superoxide dismutase and tyrosinase.[2]
- In order to function appropriately, a significant proportion of copper must be absorbed and transported to the different site of actions. Stomach and duodenum are the main sites of absorption of copper then it binds albumin and transported to different body tissues. The excess copper is excreted as feces after being a part of the bile.
- Copper is carried inside the cells by a transporter protein on the cells of the small bowel called copper membrane transporter 1 (CMT1). Others are bound to metallothionein where the rest is carried by ATOX1 to the trans-Golgi network.
- Transportation of the copper to different body tissues is regulated by ATP7B gene in the hepatocytes.
- ATP7B gene acts on transporting copper via two ways:[3][4]
- In the trans-golgi network: Formation of ceruloplasmin (which is secreted into the blood) by adding the copper to the apoceruloplasmin.
- In the cytoplasmic vesicles: Excretion of the excess copper into the bile by exocytosis against the hepatocytes memebrane.

Pathogenesis
- The following table includes the main mechanisms that play an important role in pathogenesis of Wilson’s disease.
| Impaired copper metabolism | Role in Wilson’s disease pathogenesis |
|---|---|
| Impaired copper incorporation |
|
| Copper accumulation in hepatocytes |
|
| Extrahepatic copper accumulation |
Genetics
- ATP7B gene mutation is the main cause of copper transportation impairment and Wilson’s disease.
- ATP7B gene placed on chromosome 13 and is expressed primarily in the liver, kidney, and placenta.
- Wilson’s disease ATP7B gene mutation is inhereted in an autosomal recessive pattern. The majority of the patients have no family history.
Associated conditions
- Wilson’s disease may be associated with the following conditions:[5]
Gross pathology
-
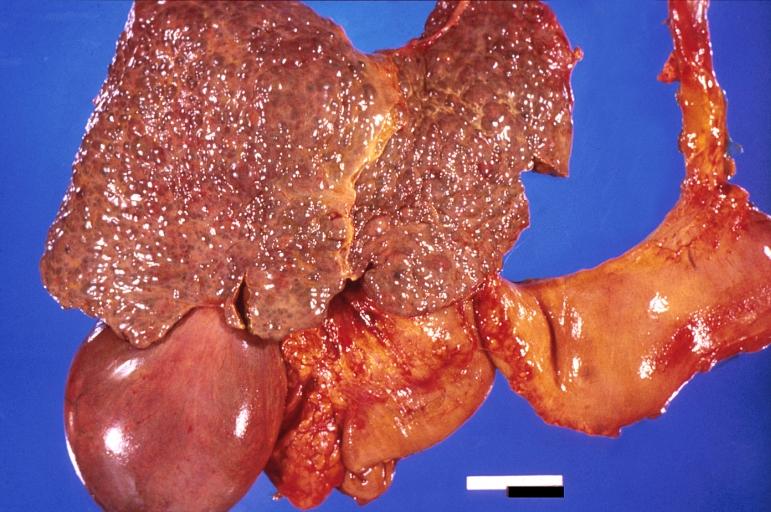 Liver cirrhosis and enlarged gall bladder in Wilson’s disease.
Liver cirrhosis and enlarged gall bladder in Wilson’s disease. -
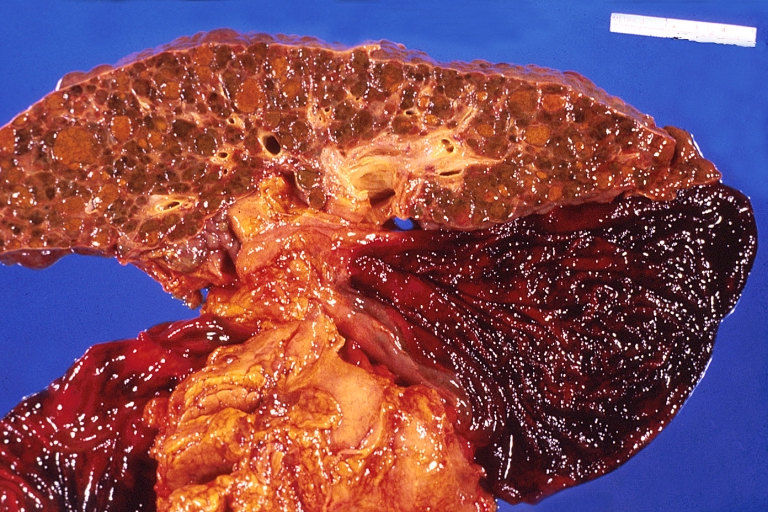 Liver cirrhosis and enlarged gall bladder in Wilson’s disease.
Liver cirrhosis and enlarged gall bladder in Wilson’s disease. -
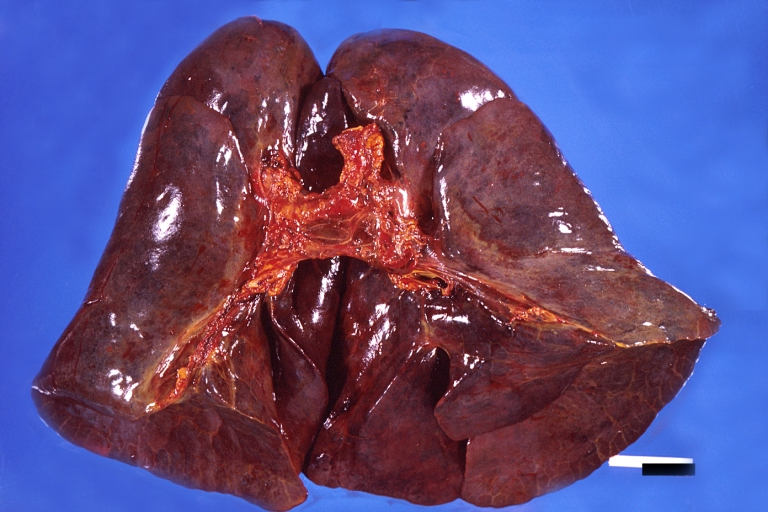 Lung, hemorrhagic bronchopneumonia, Wilson’s disease
Lung, hemorrhagic bronchopneumonia, Wilson’s disease -
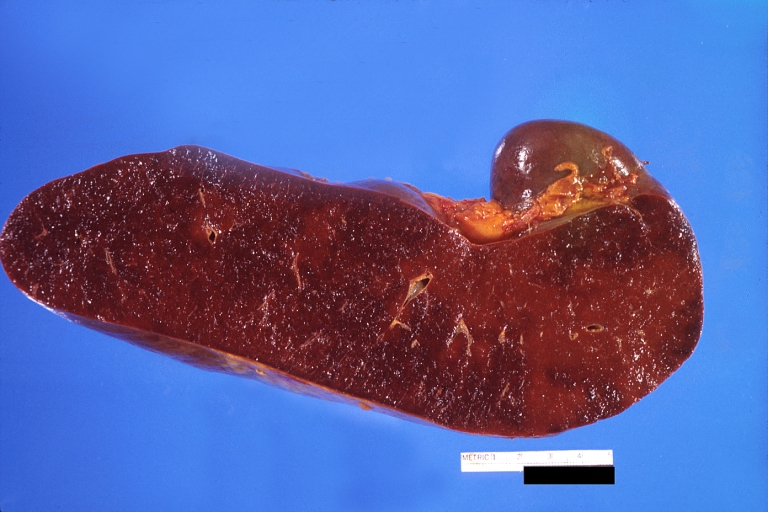 Spleen, congestion. Wilson’s disease
Spleen, congestion. Wilson’s disease




Microscopic pathology
- Histological examination of a liver biopsy may show the following:[6]
- Mild steatosis which is considered an early histological feature
- Glycogenated hepatic nuclei
- Hepatocellular necrosis
- Autoimmune hepatitis histologic features
- Fibrosis and cirrhosis (macronodular or micronodular) in advanced cases
- Fulminant liver falilure features which include:
- Hepatocellular degenration
- Parenchymal collapse
References
- ↑ Sandstead HH (1982). “Copper bioavailability and requirements”. Am J Clin Nutr. 35 (4): 809–14. PMID 6280488.
- ↑ Sandstead HH (1982). “Copper bioavailability and requirements”. Am J Clin Nutr. 35 (4): 809–14. PMID 6280488.
- ↑ Pfeiffer RF (2007). “Wilson’s Disease”. Semin Neurol. 27 (2): 123–32. doi:10.1055/s-2007-971173. PMID 17390257.
- ↑ Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD (1997). “Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae”. J Biol Chem. 272 (34): 21461–6. PMID 9261163.
- ↑ de Bie, P; Muller, P; Wijmenga, C; Klomp, L W J (2007). “Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes”. Journal of Medical Genetics. 44 (11): 673–688. doi:10.1136/jmg.2007.052746. ISSN 1468-6244.
- ↑ Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW; et al. (2006). “MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation”. AJNR Am J Neuroradiol. 27 (6): 1373–8. PMID 16775300.
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Wilson’s disease is caused by ATP7B gene mutation and impairement of copper transportation.
Causes
- Wilson’s disease is caused mainly by mutations in the genes responsible for copper transport.
- Genetic mutations:[1]
- Impairment of copper transport mechanisms include the following:[2]
- Imapired copper incorporation in ceruloplasmin
- Copper accumulation in the liver
- Extrahepatic copper accumulation
References
- ↑ Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993). “The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene”. Nat Genet. 5 (4): 327–37. doi:10.1038/ng1293-327. PMID 8298639.
- ↑ Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B; et al. (1993). “The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene”. Nat Genet. 5 (4): 344–50. doi:10.1038/ng1293-344. PMID 8298641.
Differentiating Wilson’s disease from other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Wilson’s disease must be differentiated from other diseases that cause jaundice like hemochromatosis, viral hepatitis, alcoholic hepatitis, drug induced hepatitis, and autoimmune hepatitis.
Differentiating Wilson’s disease from other diseases
The differential diagnosis for jaundice, click here.
The differential diagnosis for jaundice and RUQ pain, click here.
The differential diagnosis for jaundice and pruritis, click here.
The differential diagnosis for jaundice and fever, click here.
The differential diagnosis for jaundice, fever, and RUQ pain, click here.
The differential diagnosis for jaundice, pruritis and RUQ pain, click here.
Differential diagnosis of jaundice are: [1][2][3][4][5]
References
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Epidemiology and Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
The prevalence of Wilson’s disease is estimated to be 3 cases per 100,000 individuals worldwide. Wilson’s disease affects individuals between 5 to 35 years old.
Epidemiology and demographics
Prevalence
- The prevalence of Wilson’s disease is estimated to be 3 cases per 100,000 individuals worldwide.[1]
Age
- Wilson’s disease commonly affects inidividuals between 5 to 35 years old.
Race
- There is no racial predilection to Wilson’s disease.
Gender
- Wilson’s disease affects men and women equally.
References
- ↑ Huster D (2010). “Wilson disease”. Best Pract Res Clin Gastroenterol. 24 (5): 531–9. doi:10.1016/j.bpg.2010.07.014. PMID 20955957.
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Commpn risk factors of Wilson’s disease include newborns of disease-carrier parents. Less common risk factors inlcude excess non-vegetarian diet.
Risk factors
Common risk factors of Wilson’s disease include the following:[1]
- Newborns of parents who are carriers of the disease
Less common risk factors of Wilson’s disease include:
- Excess non-vegetarian diet
References
- ↑ Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993). “The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene”. Nat Genet. 5 (4): 327–37. doi:10.1038/ng1293-327. PMID 8298639.
Screening
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
There is insufficient evidence to recommend routine screening for Wilson’s disease.
Screening
There is insufficient evidence to recommend routine screening for Wilson’s disease.
References
Natural History, Complications and Prognosis
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
If left untreated, Wilson’s disease will lead to death as the copper accumulation in the liver and brain will lead to cirrhosis and severe dystonia respectively. Common complications of Wilson’s disease include hepatocellular carcinoma, renal failure, and persistent neurological manifestations. Prognosis of Wilson’s disease is usually good in case of early detection and proper treatment.
Natural History, Complications, and Prognosis
Natural history
- The symptoms of Wilson’s disease usually develop between age of 5 and 35 years. However, the symptoms can be presented at any age onset.
- If left untreated, Wilson’s disease will lead to death. The copper accumulation in the liver and the brain will end up with cirrhosis and severe dystonia respectively. [1]
Complications
- Common complications of Wilson’s disease include the following:
- Cirrhosis
- Hepatocellular carcinoma
- Persistent the neurological manifestations
- Renal complications as kidney stones and renal failure
- Hemolytic anemia
Prognosis
- Prognosis of Wilson’s disease is usually good with early detection and proper treatment except in the severely advanced patients.
References
- ↑ European Association for Study of Liver (2012). “EASL Clinical Practice Guidelines: Wilson’s disease”. J Hepatol. 56 (3): 671–85. doi:10.1016/j.jhep.2011.11.007. PMID 22340672.
Diagnosis
Diagnosis
Wilson’s disease diagnostic study of choice | History and Symptoms | Physical Examination | Laboratory Findings | Electrocardiogram | X-ray | CT | Echocardiography or Ultrasound | Other Imaging Findings | Other Diagnostic Studies
Treatment
Treatment
Medical Therapy | Surgery | primary prevention | secondary prevention | Cost-effectiveness of Therapy | Future or Investigational Therapies Template:Mineral metabolic pathology
ar:مرض ويلسون ca:Malaltia de Wilson de:Morbus Wilson it:Malattia di Wilson he:מחלת וילסון lb:Wilson-Krankheet hu:Wilson-kór nl:Ziekte van Wilson fi:Wilsonin tauti
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH