
Epithelioid sarcoma
For patient information, click here
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Synonyms and keywords: Epithelioid cell sarcoma; Adult epithelioid sarcoma; Epithelioid cell synovial sarcoma
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Jesus Rosario Hernandez, M.D. [2]; Ammu Susheela, M.D. [3]
Overview
Epithelioid sarcoma is a rare mesenchymal soft tissue tumor. It often occurs in the extremities of young people. Diagnosis is often delayed because of the unusual nature of the tumor. Epithelioid sarcoma was first discovered by Dr. J. Laskowski, a polish doctor, in 1961 following the description of a clinical entity called sarcoma aponeuroticum due to the involvement of aponeurosis. Enzinger coined the term epithelioid sarcoma in 1970. Mutations in the SMARCB1 gene cause epithelioid sarcoma. Epithelioid sarcoma must be differentiated from synovial sarcoma, wart, ganglion cyst, rhabdomyosarcoma, and clear cell sarcoma. Irregular (or nodular) lesions below the skin surface are the major symptom of epithelioid sarcoma. The symptoms of epithelioid sarcoma usually develop in the second decade of life and start with symptoms such as mass. Epitheliod sarcoma has a tendency for lymph node metastasis. The most common sites of epithelioid metastasis include lungs, lymph nodes, and scalp. Common complications of epithelioid sarcoma include metastasis, nerve compression, and numbness. The prognosis of epithelioid sarcoma is good for females. The predominant therapy for epithelioid sarcoma is surgical resection. Adjunctive chemotherapy, radiation, and chemoradiation may be required.
Historical Perspective
Epithelioid sarcoma was first discovered by Dr. J. Laskowski, a polish doctor, in 1961 following the description of a clinical entity called sarcoma aponeuroticum due to the involvement of aponeurosis. Enzinger coined the term epithelioid sarcoma in 1970.
Pathophysiology
Mutations in the SMARCB1 gene cause epithelioid sarcoma. On gross pathology, solid, multinodular mass, glistening gray tan appearance, and multiple areas of hemorrhage and necrosis are characteristic findings of epithelioid sarcoma. On microscopic histopathological analysis, central necrosis surrounded by bland, polygonal cells with eosinophilic cytoplasm and peripheral spindling, desmoplasia, focal calcification, or metaplastic ossification are characteristic findings of epithelioid sarcoma.
Causes
Mutations in the SMARCB1 gene cause epithelioid sarcoma.
Differentiating Epithelioid sarcoma from other Diseases
Epithelioid sarcoma must be differentiated from synovial sarcoma, wart, ganglion cysts, rhabdomyosarcoma, and clear cell sarcoma.
Epidemiology and Demographics
The epithelioid sarcoma is a very rare disease. The incidence of epithelioid sarcoma increases between ages of 10 and 39 and the average age of presentation was 27 years. Males are more commonly affected with epithelioid sarcoma than female. Epithelioid sarcoma of upper extremity usually affects individuals of the Caucasian race.
Risk Factors
The most potent risk factor in the development of epithelioid sarcoma is the presence of family history of epithelioid sarcoma.
Natural history, Complications and Prognosis
The symptoms of epithelioid sarcoma usually develop in the second decade of life and start with symptoms such as mass. Epithelioid sarcoma has a tendency to lymph node metastasis. The most common sites of epithelioid metastasis include lungs, lymph nodes, and scalp. Common complications of epithelioid sarcoma include metastasis, nerve compression, and numbness. The prognosis of epithelioid sarcoma is good for females.
Staging
The staging for epithelioid sarcoma takes into account size and location of the primary tumor, lymph node involvement, presence and location of metastasis, and histologic grade.
History and Symptoms
Irregular (or nodular) lesions below the skin surface are the major symptom of epithelioid sarcoma.
Physical Examination
Patients with epithelioid sarcoma usually appear healthy. Physical examination of patients with epithelioid sarcoma is usually remarkable for a firm to hard palpable masses in the extremities.
MRI
MRI may be performed to detect metastases of epithelioid sarcoma to determine anatomic boundaries.
Other Diagnostic Studies
Other diagnostic studies for epithelioid sarcoma include genetic testing and immunohistochemistry.
Biopsy
Tissue biopsy may be helpful in the diagnosis of epithelioid sarcoma. Findings on biopsy suggestive of epithelioid sarcoma include white nodules with infiltrating margins, epithelial cells well blended with fusiform cells with intracytoplasmic vacuoles, and “pseudo granulomatous” proliferation of cells around acellular necrotic debris.
Medical Therapy
The predominant therapy for epithelioid sarcoma is surgical resection. Adjunctive chemotherapy, radiation, and chemoradiation may be required.
Surgery
Surgery is the mainstay of treatment for epithelioid sarcoma.
References
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Epithelioid sarcoma was first discovered by Dr. J. Laskowski, a Polish physician, in 1961 following the description of a clinical entity called sarcoma aponeuroticum due to the involvement of aponeurosis. Enzinger coined the term epithelioid sarcoma in 1970.
Discovery
- Epithelioid sarcoma was first discovered by Dr. J. Laskowski, a Polish physician, in 1961 following the description of a clinical entity called sarcoma aponeuroticum due to the involvement of aponeurosis.
- In 1968, Bliss and Reed reported 4 cases of epithelioid sarcoma originating from hands and fingers sheath.
- Epithelioid sarcoma was recognized as a clinical entity only after 62 cases were reported by Enzinger in 1970. Enzinger also coined the term epithelioid sarcoma to a group of soft tissue tumors which were confused benign and malignant conditions.[1]
- In 1997, “proximal-type” of epithelioid sarcoma was described.[2]
Famous Cases
- Most significant contributions about epithelioid sarcoma were reported by Santiago et al in 1972, Soul and Enriquez in 1972, Bryan in 1974, and Prat et al in 1978.
References
- ↑ Sobanko JF, Meijer L, Nigra TP (2009). [http://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?
dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=20729965 “Epithelioid sarcoma: a review and update”] Check
|url=value (help). J Clin Aesthet Dermatol. 2 (5): 49–54. PMC 2924131. PMID 20729965. line feed character in|url=at position 54 (help) - ↑ Epithelioid sarcoma. Sarcomahelp (2016). http://sarcomahelp.org/epithelioid-sarcoma.html Accessed on February 8, 2016
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Mutations in the SMARCB1 gene cause epithelioid sarcoma. On gross pathology, a solid, multinodular mass with a glistening gray tan appearance and multiple areas of hemorrhage and necrosis are characteristic findings of epithelioid sarcoma. On microscopic histopathological analysis, central necrosis surrounded by bland, polygonal cells with eosinophilic cytoplasm and peripheral spindling, desmoplasia, focal calcification, or metaplastic ossification are characteristic findings of epithelioid sarcoma.
Pathogenesis
- Epithelioid sarcoma is the second most common soft tissue sarcoma in hand. Epithelioid sarcoma is also the sixth most common soft tissue sarcoma in the upper extremity.
- Primary site of epithelioid sarcoma is upper distal extremities. Other rare sites of epithelioid sarcoma are:
- Epithelioid sarcoma is a rare soft tissue sarcoma arising from mesenchymal tissue and characterized by epithelioid-like features.
- Epithelioid sarcoma accounts for less than 1% of all soft tissue sarcomas. Epithelioid sarcoma commonly presents itself in the distal limbs (fingers, hands, forearms, or feet) of young adults as a small, soft mass or a series of bumps. A proximal version has also been described, frequently occurring in the upper extremities.[1] Rare cases have been reported in the pelvis, vulva, penis, and spine.
Genetics
- SMARCB1 gene is involved in the pathogenesis of epithelioid sarcoma.
- The most common genetic mutation (found in 80-90% of epithelioid sarcomas) is the inactivation of the SMARCB1 gene, or the loss of INI-1 function, which is thought to be a major contributor to disease progression.[2][3] Epithelioid sarcoma typically contains chromosome 22q11.2 mutations or deletions and 8q gains, particularly i(8) (>q10). Aberrations of 18q and 8q, as well as recurrent gains at 11q13, have also been observed.[4][5][6]
- The SMARCB1 gene (also termed BAF47, INI1, or hSNF5) is located on chromosome 22q11.2[2] and codes for a member of the SWI/SNF chromatin remodeling complex. Loss of SMARCB1 function is the most common genetic mutation observed in epithelioid sarcoma, and this dysfunction is likely a major driver of disease progression. SMARCB1 is a core protein subunit of the 15 subunit SWI/SNF (or BAF) complex involved in regulating the nucleosome architecture of our genome and has been shown to be a potent tumor suppressor gene, meaning that its primary role is to control cell division and to even halt division under appropriate circumstances (i.e. signals to over-replicate).[3][7] As this tumor suppressor is commonly inactivated in epithelioid sarcoma, cell division can fail to appropriately halt, resulting in unregulated cellular growth and the formation of cancer tumors. Several research teams are currently developing techniques to reverse this loss of genetic function characteristic of epithelioid sarcoma.
Molecular Biology
VEGF
VEGF (vascular endothelial growth factor) is often over-expressed in epithelioid sarcoma.[8] This is a critical pathway in angiogenesis, a process that cancer cells use to form new blood vessels, which provide necessary elements to the tumor for tumor survival. Anti-VEGF agents such as pazopanib have shown promise across several different carcinomas and in soft tissue sarcomas.[9] In one case study, a patient with advanced metastatic vulvar epithelioid sarcoma showed a partial resolution of both lung and pleural metastases when pazopanib was administered, whereas all other therapies had failed.[10]
MET
MET (mesenchymal to epithelial transition) is another biological pathway that is likely involved in the development and progression of epithelioid sarcoma.[11][12] c-MET is a tyrosine kinase oncogene, and its signaling pathway has been implicated in a variety of malignancies, including many cancers.
Sonic hedgehog and Notch
The Sonic hedgehog and Notch signaling pathways are also suspected to be up-regulated in epithelioid sarcoma. These cell signaling pathways control cellular proliferation and differentiation. They are also involved in cancer stem cell coordination and disease invasiveness and metastasis. Hhat inhibitors (such as RU-SKI 43) block the Sonic hedgehog signaling pathway by inhibiting hedgehog palmitoyl acytl-transferase. Current trials are investigating Notch inhibitors against epithelioid sarcoma.[13]
mTOR
The frequent hyperactivation of mTOR (mammalian target of rapamycin) signaling has also been observed in epithelioid sarcoma.[12][14] The mTOR pathway has been described as a “master switch” for cellular catabolism and anabolism, and it can enhance cell cycle progression, cell survival, and block normal cell death (apoptosis).[9] Interestingly, it has been demonstrated that simply blocking mTOR signaling can result in the reactivation of the AKT pathway, negating much of the anti-mTOR’s efficacy.[12] This reactivation of AKT has been shown to be c-MET-dependent,[12] resulting in the rationale that blocking both mTOR and c-MET concurrently would show increased efficacy.
EGFR
The over-expression of epidermal growth factor receptor (EGFR) has been reported in a majority of epithelioid sarcomas.[14][15] EGFR is a member of the HER receptor family. Upon ligand binding, EGFR phosphorylation triggers the activation of downstream signaling pathways involved in critical cellular functions such as proliferation, survival, and angiogenesis.[16] In-vitro and in-vivo laboratory experiments have demonstrated that the blockade of EGFR in epithelioid sarcoma results in decreased cell proliferation, increased apoptosis, and abrogated invasion and migration capacities.[14] Of interest, while the simple blockade of EGFR with a single agent has shown limited results in the clinical setting, when used as part of a combination regime (where an EGFR inhibitor is combined with an mTOR inhibitor), a synergism has been observed, and superior tumor growth inhibition has been demonstrated.[14]
CD109
CD109 is often expressed in advanced epithelioid sarcoma and is thought to mark the cancer stem cell (or cancer initiating cell) of the disease.[17] Its level of expression has also been shown to be predictive of outcome. Cancer stem cells are a small population of tumor cells characterized by general chemo-resistance, the ability to self-renew, multi-differentiation potential, dormancy capabilities, and tumorigenesis. Therefore, cancer stem cells are thought to play key roles in the progression and relapse of cancer.
Cyclin D1
Cyclin D1 is a protein requisite for cell cycle progression and has been shown to be up-regulated in epithelioid sarcoma.[6] Cyclin D-1 is a regulator of cyclin-dependent kinases (CDK4 and CDK6). It has been shown to interact with the retinoblastoma protein (a tumor suppressor gene), CDK4 and CDK6, thyroid hormone receptor beta, and nuclear receptor coactivator 1, among others.[6] Cyclin D and CDKs promote cell cycle progression by releasing transcription factors that are important for the initiation of DNA replication. Abnormal levels of cyclin D-1 may promote rapid cell division in epithelioid sarcoma.
Gross Pathology
- Gross pathological features are:
- Solid, multinodular mass
- Glistening gray tan appearance
- Multiple areas of hemorrhage and necrosis
Microscopic Pathology
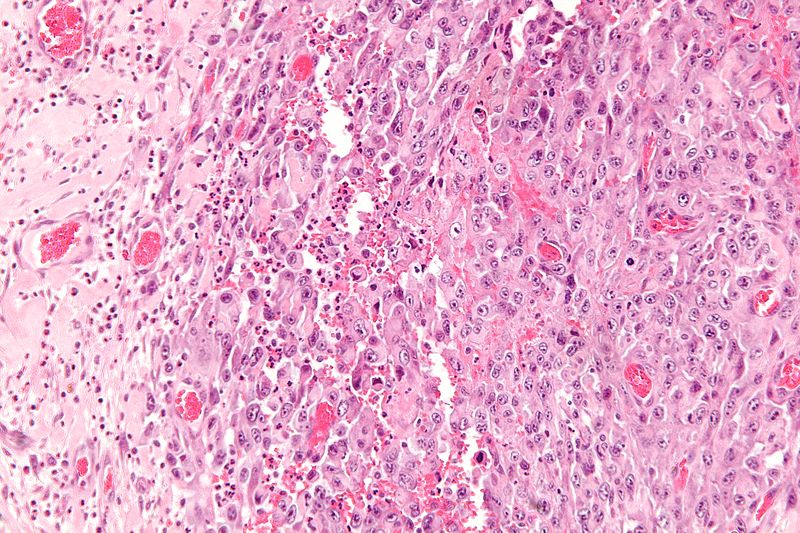
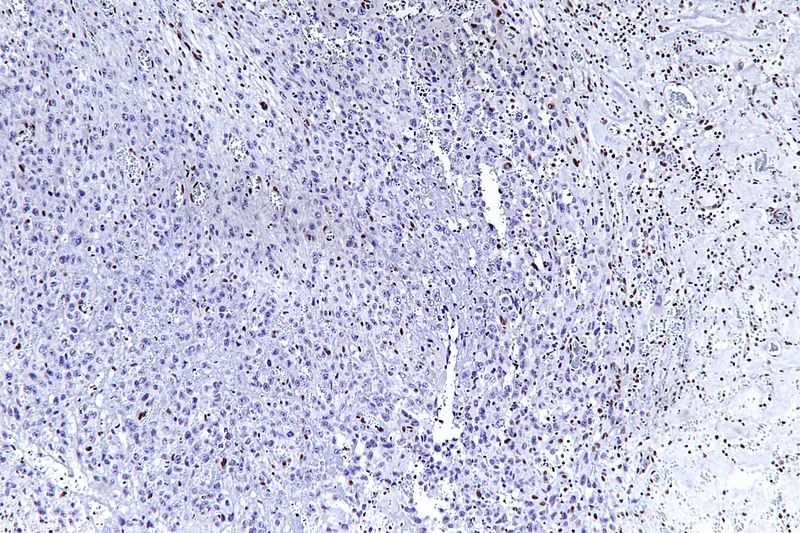
- Histologically, epithelioid sarcoma forms nodules with central necrosis surrounded by bland, polygonal cells with eosinophilic cytoplasm and peripheral spindling.[18] Epithelioid sarcomas typically express vimentin, cytokeratins, epithelial membrane antigen, and CD34, whereas they are usually negative for S100, desmin, and FLI-1.[18] They typically stain positive for CA125.[19]
- Desmoplasia, focal calcification, or metaplastic ossification may be noted.
Gallery
-
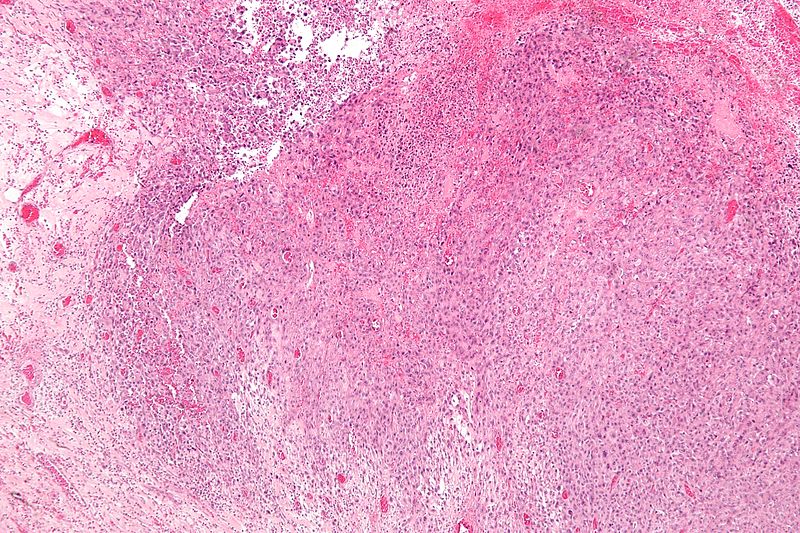 Low magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology
Low magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology -
 Low magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology
Low magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology -
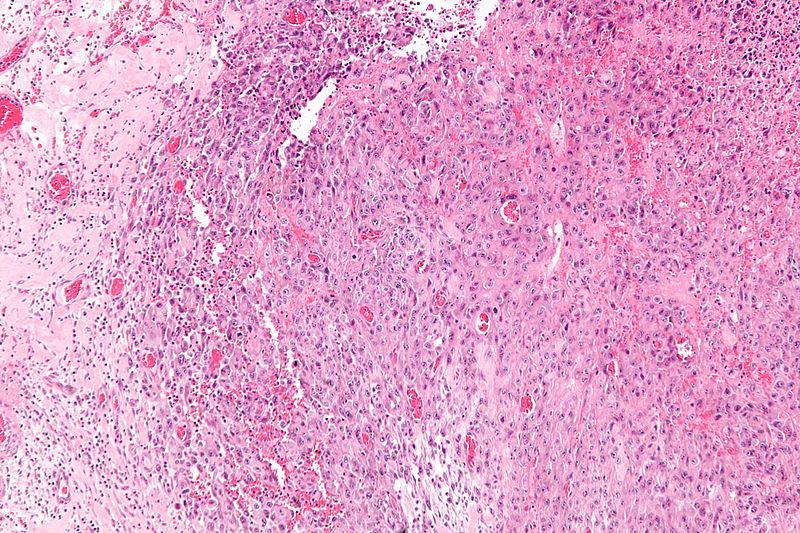 Intermediate magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology
Intermediate magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology -
 High magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology
High magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology -
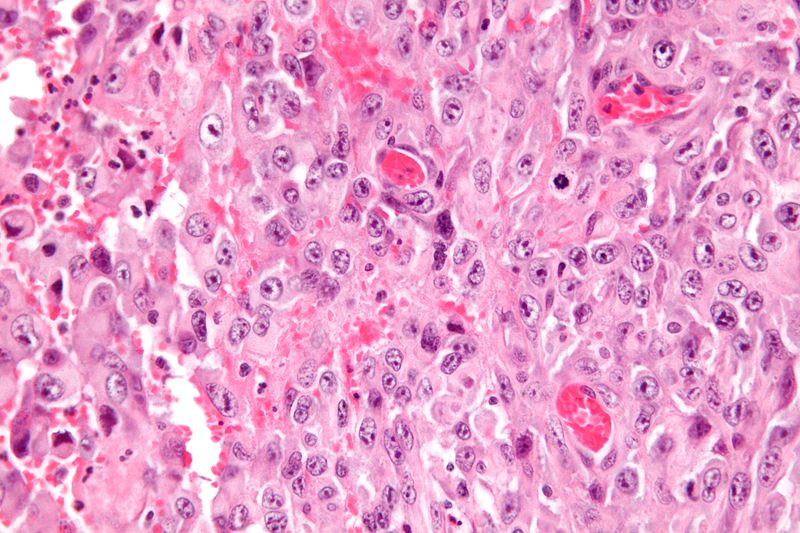 Very high magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology
Very high magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology -
Very high magnification micrograph of epithelioid sarcoma<ref> Epithelioid sarcoma librepathology





References
- ↑ Guillou, L; Wadden, C; Coindre, JM; Krausz, T; Fletcher, CD (1997). “‘Proximal-type’ epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series”. The American Journal of Surgical Pathology. 21 (2): 130–46. doi:10.1097/00000478-199702000-00002. PMID 9042279.
- ↑ 2.0 2.1 Hornick, Jason L.; Dal Cin, Paola; Fletcher, Christopher D.M. (2009). “Loss of INI1 Expression is Characteristic of Both Conventional and Proximal-type Epithelioid Sarcoma”. The American Journal of Surgical Pathology. 33 (4): 542–50. doi:10.1097/PAS.0b013e3181882c54. PMID 19033866.
- ↑ 3.0 3.1 Modena, Piergiorgio; Lualdi, Elena; Facchinetti, Federica; Galli, Lisa; Teixeira, Manuel R.; Pilotti, Silvana; Sozzi, Gabriella (2005). “SMARCB1/INI1 Tumor Suppressor Gene Is Frequently Inactivated in Epithelioid Sarcomas”. Cancer Research. 65 (10): 4012–9. doi:10.1158/0008-5472.CAN-04-3050. PMID 15899790.
- ↑ Lushnikova, Tamara; Knuutila, Sakari; Miettinen, Markku (2000). “DNA Copy Number Changes in Epithelioid Sarcoma and Its Variants: A Comparative Genomic Hybridization Study”. Modern Pathology. 13 (10): 1092–6. doi:10.1038/modpathol.3880203. PMID 11048803.
- ↑ Nishio, Jun; Iwasaki, Hiroshi; Nabeshima, Kazuki; Ishiguro, Masako; Naumann, Sabine; Isayama, Teruto; Naito, Masatoshi; Kaneko, Yasuhiko; Kikuchi, Masahiro; Bridge, Julia (2005). “Establishment of a new human epithelioid sarcoma cell line, FU-EPS-1: Molecular cytogenetic characterization by use of spectral karyotyping and comparative genomic hybridization”. International Journal of Oncology. 27 (2): 361–9. doi:10.3892/ijo.27.2.361. PMID 16010416.
- ↑ 6.0 6.1 6.2 Lin, Lin; Hicks, David; Xu, Bo; Sigel, Jessica E; Bergfeld, Wilma F; Montgomery, Elizabeth; Fisher, Cyril; Hartke, Marybeth; Tubbs, Raymond; Goldblum, John R (2005). “Expression profile and molecular genetic regulation of cyclin D1 expression in epithelioid sarcoma”. Modern Pathology. 18 (5): 705–9. doi:10.1038/modpathol.3800349. PMID 15578074.
- ↑ Kahali, Bhaskar; Yu, Jinlong; Marquez, Stefanie B.; Thompson, Kenneth W.; Liang, Shermi Y.; Lu, Li; Reisman, David (2014). “The silencing of the SWI/SNF subunit and anticancer gene BRM in Rhabdoid tumors”. Oncotarget. 5 (10): 3316–32. doi:10.18632/oncotarget.1945. PMC 4102812. PMID 24913006.
- ↑ Kuhnen, Cornelius; Lehnhardt, Marcus; Tolnay, Edina; Muehlberger, Thomas; Vogt, Peter M.; Müller, Klaus-Michael (2000). “Patterns of expression and secretion of vascular endothelial growth factor in malignant soft-tissue tumours”. Journal of Cancer Research and Clinical Oncology. 126 (4): 219–25. doi:10.1007/s004320050036. PMID 10782895.
- ↑ 9.0 9.1 Martín Liberal, Juan; Lagares-Tena, Laura; Sáinz-Jaspeado, Miguel; Mateo-Lozano, Silvia; García del Muro, Xavier; Tirado, Oscar M. (2012). “Targeted Therapies in Sarcomas: Challenging the Challenge”. Sarcoma. 2012: 626094. doi:10.1155/2012/626094. PMC 3372278. PMID 22701332.
- ↑ Chung, Hye Won. “The treatment of pazopanib on vulvar epithelioid sarcoma: A case report and review of literature”.
- ↑ Kuhnen, C.; Tolnay, Edina; Steinau, Hans Ulrich; Voss, Bruno; Müller, Klaus-Michael (1998). “Expression of c-Met receptor and hepatocyte growth factor/scatter factor in synovial sarcoma and epithelioid sarcoma”. Virchows Archiv. 432 (4): 337–42. doi:10.1007/s004280050175. PMID 9565343.
- ↑ 12.0 12.1 12.2 12.3 Imura, Yoshinori; Yasui, Hirohiko; Outani, Hidetatsu; Wakamatsu, Toru; Hamada, Kenichiro; Nakai, Takaaki; Yamada, Shutaro; Myoui, Akira; Araki, Nobuhito; Ueda, Takafumi; Itoh, Kazuyuki; Yoshikawa, Hideki; Naka, Norifumi (2014). “Combined targeting of mTOR and c-MET signaling pathways for effective management of epithelioid sarcoma”. Molecular Cancer. 13: 185. doi:10.1186/1476-4598-13-185. PMC 4249599. PMID 25098767.
- ↑ Clinical trial number NCT01154452 for “Vismodegib and Gamma-Secretase/Notch Signalling Pathway Inhibitor RO4929097 in Treating Patients With Advanced or Metastatic Sarcoma” at ClinicalTrials.gov
- ↑ 14.0 14.1 14.2 14.3 Xie, X.; Ghadimi, M. P. H.; Young, E. D.; Belousov, R.; Zhu, Q.-s.; Liu, J.; Lopez, G.; Colombo, C.; Peng, T.; Reynoso, D.; Hornick, J. L.; Lazar, A. J.; Lev, D. (2011). “Combining EGFR and mTOR Blockade for the Treatment of Epithelioid Sarcoma”. Clinical Cancer Research. 17 (18): 5901–12. doi:10.1158/1078-0432.CCR-11-0660. PMC 3176924. PMID 21821699.
- ↑ Cascio, Michael J; O’Donnell, Richard J; Horvai, Andrew E (2010). “Epithelioid sarcoma expresses epidermal growth factor receptor but gene amplification and kinase domain mutations are rare”. Modern Pathology. 23 (4): 574–80. doi:10.1038/modpathol.2010.2. PMID 20118913.
- ↑ Yang, J.-L.; Hannan, M.T.; Russell, P.J.; Crowe, P.J. (2006). “Expression of HER1/EGFR protein in human soft tissue sarcomas”. European Journal of Surgical Oncology. 32 (4): 466–8. doi:10.1016/j.ejso.2006.01.012. PMID 16524687.
- ↑ Ahmad, Aamir; Emori, Makoto; Tsukahara, Tomohide; Murase, Masaki; Kano, Masanobu; Murata, Kenji; Takahashi, Akari; Kubo, Terufumi; Asanuma, Hiroko; Yasuda, Kazuyo; Kochin, Vitaly; Kaya, Mitsunori; Nagoya, Satoshi; Nishio, Jun; Iwasaki, Hiroshi; Sonoda, Tomoko; Hasegawa, Tadashi; Torigoe, Toshihiko; Wada, Takuro; Yamashita, Toshihiko; Sato, Noriyuki (2013). “High Expression of CD109 Antigen Regulates the Phenotype of Cancer Stem-Like Cells/Cancer-Initiating Cells in the Novel Epithelioid Sarcoma Cell Line ESX and Is Related to Poor Prognosis of Soft Tissue Sarcoma”. PLoS ONE. 8 (12): e84187. doi:10.1371/journal.pone.0084187. PMC 3869840. PMID 24376795.
- ↑ 18.0 18.1 Armah, Henry B. Armah; Parwani, Anil V. (2009). “Epithelioid sarcoma”. Archives of Pathology & Laboratory Medicine. 133 (5): 814–9. doi:10.1043/1543-2165-133.5.814 (inactive October 6, 2015). PMID 19415960.
- ↑ Kato, Hiroshi; Hatori, Masahito; Kokubun, Shoichi; Watanabe, Mika; Smith, Richard A; Hotta, Tetsuo; Ogose, Akira; Morita, Tetsuro; Murakami, Takashi; Aiba, Setsuya (2004). “CA125 expression in epithelioid sarcoma”. Japanese Journal of Clinical Oncology. 34 (3): 149–54. doi:10.1093/jjco/hyh027. PMID 15078911.
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Mutations in the SMARCB1 gene cause epithelioid sarcoma.
Causes
Mutations in the SMARCB1 gene cause epithelioid sarcoma.[1]
Reference
- ↑ Epithelioid sarcoma. Sarcomahelp (2016). http://sarcomahelp.org/epithelioid-sarcoma.html Accessed on February 8, 2016
Differentiating Epithelioid sarcoma from other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Epithelioid sarcoma must be differentiated from synovial sarcoma, wart, ganglion cysts, rhabdomyosarcoma, and clear cell sarcoma.
Differentiating Epithelioid sarcoma from other Diseases
- Synovial sarcoma
- Ulcerating squamous cell carcinoma[1]
- Granulomatous diseases
- Traumatic wound
- Wart
- Ganglion cysts
- Giant cell tumors
- Rhabdomyosarcoma
- Clear cell sarcoma
- Vascular sarcoma
| Keratin | CD34 | Cytoplasm | In-sutu Kerataniztion | Location | Age group | H/O of epithelial injury | CA125 | CK5/6 | GCDFP15, S100, actin | Necrosis | Calcification | Hemangiopericytomatous vessels | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Epitheloid Sarcoma | Positive | Positive | Sharply defined | Negative | Hand and wrist | Under 40 years | No | Positive | Rare, Local | Negative | Extensive | Infrequent | Negative |
| Palisaded Necrotic Granuloma | Neagtive | Negative | Indistinct | Uncommon | |||||||||
| Squamous Carcinoma | Negative | Positive | >40 | Positive | Negative in cutaneous SCC
Positive everywhere |
Extensive | |||||||
| Sweat Gland Carcinoma | Negative | Positive | |||||||||||
| Monophasic Synovial Sarcoma | Negative | Negative | Focal | Frequent | Positive | ||||||||
| Melanoma | Negative | Negative | Negative | Positive | |||||||||
| Epithelioid Hemangioendothelioma | Negative | Uncommon on extremities | |||||||||||
| Sclerosing Epithelioid Fibrosarcoma | Negative | Cords of cells in hylainzed stroma | |||||||||||
| Ischemic Fasciitis | Negative | Basophilic or amphophilic |
| Disease | History/demography | Symptoms | Physical examination | Diagnosis | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Palpable mass | Pain | Others | Mass tenderness | Others | Genetics | Imaging | Histology | |||
| Epitheloid Sarcoma |
|
+ | – | Mainly cutaneous manifestations | – |
|
|
|
| |
| Rhabdomyosarcoma[2][3][4][5] |
|
+ | + |
|
+/- |
Mutations in: |
CT scan:
MRI:
|
| ||
| Wilms tumor[6][7][8][9][10] |
|
+ | + |
|
+/- | Present mutations of: | Ultrasound:
|
| ||
| Ewing sarcoma[11][12][13][14] |
|
+ | + | + |
|
Radiographic of region:
MRI:
|
| |||
| Pediatric neuroblastoma [15][16][17][18] |
Age distribution:
|
+ (Abdominal) |
+ |
+(Abdominal) |
CT scan:
MRI:
|
| ||||
| Pediatric pheochromocytoma[19][20][21][22] |
|
– | +/- | – | Genetic mutation in: | Ultrasound:
|
Positive stains for:
| |||
| Pediatric osteosarcoma[23][24][25] |
|
+ | + | + |
|
|
Radiography:
MRI: |
| ||
| Pediatric liposarcoma[26][27][28][29] |
|
+ | +/- | – |
|
|
CT scan:
MRI: |
Divided into following subtypes:
Common findings:
| ||
| Pediatric acute myelocytic leukemia[30][31][32][33] |
|
+/- ( Abdominal mass, mediastinal mass) | + (bone pain, joint pain) | +/- | Genetic translocations include:
|
Radiography:
|
| |||
| Pediatric acute lymphoblastic leukemia[34][35] |
|
+/- (Musculoskeletal pain) | – | Chromosomal translocations:
|
Radiography:
|
Divided into 3 subgroups:
L1:
L2:
L3:
| ||||
| Pediatric non-hodgkin lymphoma[36][37][38] |
|
+ | – | + (Chest tenderness) | Radiography:
|
Histology findings of non-hodgkin lymphoma depend on: | ||||
References
- ↑ Epithelioid sarcoma. Sarcomahelp (2016). http://sarcomahelp.org/epithelioid-sarcoma.html Accessed on February 8, 2016
- ↑ Egas-Bejar D, Huh WW (2014). “Rhabdomyosarcoma in adolescent and young adult patients: current perspectives”. Adolesc Health Med Ther. 5: 115–25. doi:10.2147/AHMT.S44582. PMC 4069040. PMID 24966711.
- ↑ Dasgupta R, Fuchs J, Rodeberg D (2016). “Rhabdomyosarcoma”. Semin Pediatr Surg. 25 (5): 276–283. doi:10.1053/j.sempedsurg.2016.09.011. PMID 27955730.
- ↑ Park K, van Rijn R, McHugh K (2008). “The role of radiology in paediatric soft tissue sarcomas”. Cancer Imaging. 8: 102–15. doi:10.1102/1470-7330.2008.0014. PMC 2365455. PMID 18442956.
- ↑ Shern JF, Yohe ME, Khan J (2015). “Pediatric Rhabdomyosarcoma”. Crit Rev Oncog. 20 (3–4): 227–43. PMC 5486973. PMID 26349418.
- ↑ Hartman DS, Sanders RC (April 1982). “Wilms’ tumor versus neuroblastoma: usefulness of ultrasound in differentiation”. J Ultrasound Med. 1 (3): 117–22. PMID 6152936.
- ↑ De Campo JF (1986). “Ultrasound of Wilms’ tumor”. Pediatr Radiol. 16 (1): 21–4. PMID 3003660.
- ↑ Cahan LD (1985). “Failure of encephalo-duro-arterio-synangiosis procedure in moyamoya disease”. Pediatr Neurosci. 12 (1): 58–62. PMID 4080660.
- ↑ Coppes MJ, Pritchard-Jones K (2000). “Principles of Wilms’ tumor biology”. Urol Clin North Am. 27 (3): 423–33, viii. PMID 10985142.
- ↑ Davidoff AM (2012). “Wilms tumor”. Adv Pediatr. 59 (1): 247–67. doi:10.1016/j.yapd.2012.04.001. PMC 3589819. PMID 22789581.
- ↑ Burchill SA (2003). “Ewing’s sarcoma: diagnostic, prognostic, and therapeutic implications of molecular abnormalities”. J Clin Pathol. 56 (2): 96–102. PMC 1769883. PMID 12560386.
- ↑ Maygarden SJ, Askin FB, Siegal GP, Gilula LA, Schoppe J, Foulkes M; et al. (1993). “Ewing sarcoma of bone in infants and toddlers. A clinicopathologic report from the Intergroup Ewing’s Study”. Cancer. 71 (6): 2109–18. PMID 8443760.
- ↑ Panicek DM, Gatsonis C, Rosenthal DI, Seeger LL, Huvos AG, Moore SG; et al. (1997). “CT and MR imaging in the local staging of primary malignant musculoskeletal neoplasms: Report of the Radiology Diagnostic Oncology Group”. Radiology. 202 (1): 237–46. doi:10.1148/radiology.202.1.8988217. PMID 8988217.
- ↑ Grünewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Álava E, Kovar H; et al. (2018). “Ewing sarcoma”. Nat Rev Dis Primers. 4 (1): 5. doi:10.1038/s41572-018-0003-x. PMID 29977059.
- ↑ Lonergan GJ, Schwab CM, Suarez ES, Carlson CL (2002). “Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: radiologic-pathologic correlation”. Radiographics. 22 (4): 911–34. doi:10.1148/radiographics.22.4.g02jl15911. PMID 12110723.
- ↑ Golden CB, Feusner JH (2002). “Malignant abdominal masses in children: quick guide to evaluation and diagnosis”. Pediatr Clin North Am. 49 (6): 1369–92, viii. PMID 12580370.
- ↑ Angstman KB, Miser JS, Franz WB (1990). “Neuroblastoma”. Am Fam Physician. 41 (1): 238–44. PMID 2403727.
- ↑ Musarella MA, Chan HS, DeBoer G, Gallie BL (1984). “Ocular involvement in neuroblastoma: prognostic implications”. Ophthalmology. 91 (8): 936–40. PMID 6493702.
- ↑ Leung K, Stamm M, Raja A, Low G (2013). “Pheochromocytoma: the range of appearances on ultrasound, CT, MRI, and functional imaging”. AJR Am J Roentgenol. 200 (2): 370–8. doi:10.2214/AJR.12.9126. PMID 23345359.
- ↑ Stein PP, Black HR (1991). “A simplified diagnostic approach to pheochromocytoma. A review of the literature and report of one institution’s experience”. Medicine (Baltimore). 70 (1): 46–66. PMID 1988766.
- ↑ Bravo EL (1991). “Pheochromocytoma: new concepts and future trends”. Kidney Int. 40 (3): 544–56. PMID 1787652.
- ↑ Bravo EL (1991). “Pheochromocytoma: new concepts and future trends”. Kidney Int. 40 (3): 544–56. PMID 1787652.
- ↑ Dorfman HD, Czerniak B (1995). “Bone cancers”. Cancer. 75 (1 Suppl): 203–10. PMID 8000997.
- ↑ Yarmish G, Klein MJ, Landa J, Lefkowitz RA, Hwang S (2010). “Imaging characteristics of primary osteosarcoma: nonconventional subtypes”. Radiographics. 30 (6): 1653–72. doi:10.1148/rg.306105524. PMID 21071381.
- ↑ Araki N, Uchida A, Kimura T, Yoshikawa H, Aoki Y, Ueda T; et al. (1991). “Involvement of the retinoblastoma gene in primary osteosarcomas and other bone and soft-tissue tumors”. Clin Orthop Relat Res (270): 271–7. PMID 1884549.
- ↑ Shmookler BM, Enzinger FM (1983). “Liposarcoma occurring in children. An analysis of 17 cases and review of the literature”. Cancer. 52 (3): 567–74. PMID 6861094.
- ↑ Marcus KC, Grier HE, Shamberger RC, Gebhardt MC, Perez-Atayde A, Silver B; et al. (1997). “Childhood soft tissue sarcoma: a 20-year experience”. J Pediatr. 131 (4): 603–7. PMID 9386667.
- ↑ Murphey MD, Arcara LK, Fanburg-Smith J (2005). “From the archives of the AFIP: imaging of musculoskeletal liposarcoma with radiologic-pathologic correlation”. Radiographics. 25 (5): 1371–95. doi:10.1148/rg.255055106. PMID 16160117.
- ↑ Italiano A, Cardot N, Dupré F, Monticelli I, Keslair F, Piche M; et al. (2007). “Gains and complex rearrangements of the 12q13-15 chromosomal region in ordinary lipomas: the “missing link” between lipomas and liposarcomas?”. Int J Cancer. 121 (2): 308–15. doi:10.1002/ijc.22685. PMID 17372913.
- ↑ Yamamoto JF, Goodman MT (2008). “Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997-2002”. Cancer Causes Control. 19 (4): 379–90. doi:10.1007/s10552-007-9097-2. PMID 18064533.
- ↑ Cancer Genome Atlas Research Network. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ; et al. (2013). “Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia”. N Engl J Med. 368 (22): 2059–74. doi:10.1056/NEJMoa1301689. PMC 3767041. PMID 23634996.
- ↑ Islam A, Catovsky D, Goldman JM, Galton DA (1985). “Bone marrow biopsy changes in acute myeloid leukaemia. I: Observations before chemotherapy”. Histopathology. 9 (9): 939–57. PMID 3864727.
- ↑ Orazi A (2007). “Histopathology in the diagnosis and classification of acute myeloid leukemia, myelodysplastic syndromes, and myelodysplastic/myeloproliferative diseases”. Pathobiology. 74 (2): 97–114. doi:10.1159/000101709. PMID 17587881.
- ↑ Zuckerman T, Rowe JM (2014). “Pathogenesis and prognostication in acute lymphoblastic leukemia”. F1000Prime Rep. 6: 59. doi:10.12703/P6-59. PMC 4108947. PMID 25184049.
- ↑ Pui CH, Robison LL, Look AT (2008). “Acute lymphoblastic leukaemia”. Lancet. 371 (9617): 1030–43. doi:10.1016/S0140-6736(08)60457-2. PMID 18358930.
- ↑ Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL; et al. (2013). “Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma”. Blood. 121 (9): 1604–11. doi:10.1182/blood-2012-09-457283. PMC 3587323. PMID 23297126.
- ↑ Sandlund JT (2015). “Non-Hodgkin Lymphoma in Children”. Curr Hematol Malig Rep. 10 (3): 237–43. doi:10.1007/s11899-015-0277-y. PMID 26174528.
- ↑ El-Galaly TC, Hutchings M (2015). “Imaging of non-Hodgkin lymphomas: diagnosis and response-adapted strategies”. Cancer Treat Res. 165: 125–46. doi:10.1007/978-3-319-13150-4_5. PMID 25655608.
Epidemiology and Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Epithelioid sarcoma is a very rare disease. The incidence of epithelioid sarcoma increases between ages of 10 and 39 and the average age at presentation is 27 years. Males are more commonly affected with epithelioid sarcoma than females. Epithelioid sarcoma of the upper extremity usually affects individuals of the Caucasian race.
Epidemiology and Demographics
Age
- The incidence of epithelioid sarcoma increases between ages of 10 and 39 and the average age at presentation is 27 years.[1]
Gender
- Males are more commonly affected with epithelioid sarcoma than females.[2] The male to female ratio is approximately 1.8:1.
Race
- Epithelioid sarcoma of upper extremity usually affects individuals of the Caucasian race.
References
- ↑ Jacobs AJ, Michels R, Stein J, Levin AS (2015). “Improvement in Overall Survival from Extremity Soft Tissue Sarcoma over Twenty Years”. Sarcoma. 2015: 279601. doi:10.1155/2015/279601. PMC 4363656. PMID 25821397.
- ↑ Archer, I; Brown, R; Fitton, J (1984). “Epithelioid sarcoma in the hand”. The Journal of Hand Surgery: British & European Volume. 9 (2): 207–209. doi:10.1016/S0266-7681(84)80035-2. ISSN 0266-7681.
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Epithelioid sarcoma is a very rare disease. The incidence of epithelioid sarcoma increases between ages of 10 and 39 and the average age at presentation is 27 years. Males are more commonly affected with epithelioid sarcoma than females. Epithelioid sarcoma of the upper extremity usually affects individuals of the Caucasian race.
Epidemiology and Demographics
Age
- The incidence of epithelioid sarcoma increases between ages of 10 and 39 and the average age at presentation is 27 years.[1]
Gender
- Males are more commonly affected with epithelioid sarcoma than females.[2] The male to female ratio is approximately 1.8:1.
Race
- Epithelioid sarcoma of upper extremity usually affects individuals of the Caucasian race.
References
- ↑ Jacobs AJ, Michels R, Stein J, Levin AS (2015). “Improvement in Overall Survival from Extremity Soft Tissue Sarcoma over Twenty Years”. Sarcoma. 2015: 279601. doi:10.1155/2015/279601. PMC 4363656. PMID 25821397.
- ↑ Archer, I; Brown, R; Fitton, J (1984). “Epithelioid sarcoma in the hand”. The Journal of Hand Surgery: British & European Volume. 9 (2): 207–209. doi:10.1016/S0266-7681(84)80035-2. ISSN 0266-7681.
Natural History, Complications and Prognosis
Please help WikiDoc by adding content here. It’s easy! Click here to learn about editing.
References
complications and prognosis
Diagnosis
Diagnosis
Staging | History and Symptoms | Physical Examination | MRI | Other Diagnostic Studies | Biopsy
Treatment
Treatment
Medical Therapy | Surgery | Cost-Effectiveness of Therapy | Future or Investigational Therapies
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH