
Pheochromocytoma
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Synonyms and keywords: Phaeochromocytoma; Pheochromocytoma; Chromaffin paraganglioma; Chromaffin tumor; Chromaffinoma
For patient information click here
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
Pheochromocytoma is a neuroendocrine tumor of the medulla of the adrenal glands and extra-adrenal chromaffin tissue, which fail to involute after birth, they secrete excessive amounts of catecholamines, usually epinephrine and norepinephrine. Extra-adrenal paragangliomas (often described as extra-adrenal pheochromocytomas) are closely related, though less common. Pheochromocytoma originates from the chromaffin cells of the sympathetic nervous system ganglia and is named based upon the primary anatomical site of origin. The incidence of pheochromocytoma ranges from a low of 0.2 per 100,000 persons to a high of 0.8 per 100,000 persons. The average age at diagnosis is 24.9 years in hereditary cases and 43.9 years in sporadic cases with men and women are equally affected. MRI and CT scan are used for the diagnosis of pheochromocytoma. Surgery is the mainstay of the treatment.
Historical Perspective
In 1886, Fränkel made the first description of a patient with pheochromocytoma. In 1912, Ludwig Pick formulated the term pheochromocytoma.1912. In 1926, the first surgical removal of pheochromocytoma in the Military Medical Academy in Yugoslavia was performed by Professor Isidor Papo.
Pathophysiology
Pheochromocytoma arises from chromaffin cells of the adrenal medulla.On gross pathology, pheochromocytoma has a multinodular and a multicentric pattern of growth. On microscopic histopathological analysis, nesting (Zellballen) pattern composed of well-defined clusters of tumor cells separated by fibrovascular stroma is a characteristic finding. It may be benign or malignant, familial origin(multiple endocrine neoplasia type 2) or sporadic one. Both of them have genetic origin depends on a large number of genes: VHL, SDH, NF1, RET.
Classification
Pheochromocytoma may be classified based on nature of the tumor into benign and malignant. It can also be classified based on spread into local, regional, and metastatic.Another classification based on origin divides pheochromocytoma into familial, non-familial and sporadic forms.
Causes
Pheochromocytoma develops in called chromaffin cells, found in adrenal medulla which secretes adrenaline, noradrenaline, and dopamine. The genetic base of pheochromocytoma depends on 2 clusters: cluster 1 tumors are noradrenergic. Cluster 2 tumors are adrenergic. Familial pheochromocytoma may be caused by a mutation of either SDHD, VHL, SDHB, RET, NF1 genes.
Differentiating Pheochromocytoma from other Diseases
Pheochromocytoma must be differentiated from other causes of paroxysmal hypertension including severe paroxysmal hypertension (Pseudopheochromocytoma), panic disorder, Factitious hypertension, carcinoid syndrome, Migraine headache, Hyperthyroidism, Renovascular hypertension, Hypoglycemia, Labile hypertension (White coat hypertension), Stroke and compression of the lateral medulla, Seizures, Baroreflex failure, and drugs.
Epidemiology and Demographics
The incidence of pheochromocytoma ranges from a low of 0.2 per 100,000 persons to a high of 0.8 per 100,000 persons. The average age at diagnosis is 24.9 years in hereditary cases and 43.9 years in sporadic cases with men and women equally affected.
Risk Factors
Pheochromocytoma is more common in third decade of life, people with family history of multiple endocrine neoplasias, Von Hippel-Lindau disease, neurofibromatosis type 1, hereditary paraganglioma syndromes.
Screening
Familial pheochromocytoma is associated with multiple endocrine neoplasias, VHL and neurofibromatosis1 and should be screened by plasma fractionated metanephrines levels. The next step is to obtain 24-hour urinaryfractionated metanephrine levels. Imaging should be considered if the initial tests are positive. Genetic testing also should be performed in high-risk patients
Natural History, Complications and Prognosis
Pheochromocytoma is an adrenaline-secreting tumor, usually develop in the fifth decade of life. Symptoms start with tachycardia, hypertension, headache, and sweating. A massive release of catecholamines can cause hyperglycemia, malignant hypertension, and metastasis. The prognosis of pheochromocytoma is generally good but metastatic pheochromocytoma has a 5-year survival rate of approximately 50%.
Diagnosis
History and Symptoms
Symptoms of pheochromocytoma include palpitations, anxiety, and headaches.
Physical Examination
Common physical exam findings of pheochromocytoma include tachycardia, hypertension, and orthostatic hypotension.
Laboratory Findings
Laboratory findings consistent with the diagnosis of pheochromocytoma include elevated catecholamines and metanephrine levels.
Electrocardiogram
On EKG, pheochromocytoma is characterized by the presence of sinus tachycardia and supraventricular tachycardia.
X-ray
There are no x-ray findings associated with pheochromocytoma.
CT
Head, neck, chest, and abdominal CT scans may be helpful in the diagnosis of pheochromocytoma.
MRI
Head, neck, chest, and abdominal MRI may be helpful in the diagnosis of pheochromocytoma.
Other Imaging Findings
123I-metaiodobenzylguanidine (MIBG) scintigraphy coupled with CT scan imaging can be used for diagnosis of pheochromocytoma.
Other Diagnostic Studies
Clonidine suppression test may be used in the diagnosis of pheochromocytoma.
Treatment
Medical Therapy
Treatment with alpha-blockers (example: phenoxybenzamine) followed by beta-blockers (example: atenolol) is required before surgery. Other drugs can be used such as calcium channel blockers and metyrosine. Adjunctive chemotherapy and radiation are used in metastatic disease. Hypertensive crisis can be managed by using sodium nitroprusside, phentolamine, and nicardipine.
Surgery
Surgery is the mainstay of treatment for pheochromocytoma. Adrenalectomy; Laparoscopic transabdominal and retroperitoneal approaches have been used successfully for nonmetastatic abdominal pheochromocytomas. The patient should receive glucocorticoid stress coverage in bilateral adrenalectomy.
Primary Prevention
Biochemical screening for family members of MEN2 patients is mandatory. Genetic testing should be performed in first-degree relatives of a patient with proven germline RET mutation.
Secondary Prevention
Preoperative treatment of pheochromocytoma is the best way to reduce complications and postoperative follow up is the best way to reduce recurrence.
References
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Mohammed Abdelwahed M.D[2] Ifrah Fatima, M.B.B.S[3]
Overview
Adrenal pheochromocytoma and its typical clinical presentation was first described by Frankel in 1886. The term pheochromocytoma was coined by Ludwig Pick in 1912. In 1926, Cesar Roux in Switzerland, Charles H. Mayo in the United States, and Isidor Papo in Yugoslavia were the first surgeons to successfully remove pheochromocytomas. An autopsy revealed President Eisenhower had a 1.5-cm pheochromocytoma in the left adrenal gland.
Historical Perspective
Discovery
- Adrenal pheochromocytoma and its typical clinical presentation was first described by Charles Sugrue in 1800.
- Its histological findings were first reported by Felix Fraenkel and Max Schottelius in 1886.
- Manasse in 1893 reported four cases of adrenocortical tumors and one case of an adrenal medulla tumor.
- Extra-adrenal pheochromocytoma was first described by Alezais and Peyron in 1908.
- The term pheochromocytoma was coined by Ludwig Pick in 1912.
- In 1922, the association between paroxysmal hypertension and pheochromocytoma was described by L’Abbe et al.
- In the early 1900s, there were case reports describing genetic implications in the pathogenesis of pheochromocytomas and its association with syndromes like NF1, VHL, and multiple endocrine neoplasia type 2 (MEN2) emerged.
- Later between 1940 and 1960, literature reviews and case series defined the genetic associations. [1]
Landmark Events in the Development of Treatment Strategies
- In 1926, Cesar Roux in Switzerland, Charles H. Mayo in the United States, and Isidor Papo in Yugoslavia were the first surgeons to successfully surgically remove pheochromocytomas. [2] [3]
Famous Cases
The following are a few famous cases of pheochromocytoma:
- President Eisenhower– After his death, an autopsy revealed a 1.5-cm pheochromocytoma in the left adrenal gland. On analysis of his blood pressure through his life, fluctuating systolic and diastolic blood pressure spikes were documented. [4]
References
- ↑ Else T (2015). “15 YEARS OF PARAGANGLIOMA: Pheochromocytoma, paraganglioma and genetic syndromes: a historical perspective”. Endocr Relat Cancer. 22 (4): T147–59. doi:10.1530/ERC-15-0221. PMID 26273101.
- ↑ Kantorovich V, Pacak K (2010). “Pheochromocytoma and paraganglioma”. Prog Brain Res. 182: 343–73. doi:10.1016/S0079-6123(10)82015-1. PMC 4714594. PMID 20541673.
- ↑ Kudva YC, Sawka AM, Young WF (2003). “Clinical review 164: The laboratory diagnosis of adrenal pheochromocytoma: the Mayo Clinic experience”. J Clin Endocrinol Metab. 88 (10): 4533–9. doi:10.1210/jc.2003-030720. PMID 14557417.
- ↑ Schmoldt A, Benthe HF, Haberland G (1975). “Digitoxin metabolism by rat liver microsomes”. Biochem Pharmacol. 24 (17): 1639–41. PMID doi.org/10.1016/j.amjcard.2006.12.043 Check
|pmid=value (help).
Classification
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
Pheochromocytomas and paragangliomas (collectively referred to as PPGLs) are rare tumors that originate from chromaffin cells in the adrenal medulla (pheochromocytoma) or in the extra-adrenal neural ganglia (paraganglioma). These tumors can be either biochemically active (producing a catecholamine like epinephrine, nor-epinephrine and dopamine) or biochemically silent. PPGLs can be either sporadic or genetic, with association to several familial syndromes. PPGLs can also be classified according to their spread into local, regional, or metastatic. The defining characteristic of malignancy in PPGLs is the presence of metastasis.
Classification
Pheochromocytoma and paragangliomas may be classified into several subtypes based on:
- Type of cells
- Nature of tumor
- Location
- Biochemical secretory patterns
- Genetics
- Mutations and pathogenetic pathways
Classification based on type of cells the tumor is derived from
Pheochromocytomas and paragangliomas may be classified according to the cells they are derived from: [1]
- Sympathetic– adrenal medulla or sympathetic trunk
- Parasympathetic– carotid body, glomus tympanicum, glomus jugulare, glomus vagale.
Classification based on nature of tumor
Malignant and benign tumors share the same biochemical and histological characters. The only difference is the ability of the malignant tumor to metastasize to distant tissues and have high rates of recurrence.
- According to the WHO 2017 Classification of Tumors of Endocrine Organs, all parangangliomas have metastatic potential and hence the term “malignant” was replaced with “metastatic”. [2]
- Common sites of metastasis include:
Classification based on location
- 95% of pheochromocytomas are found in the abdomen
- Intra-adrenal– 85-90%
- Extra-adrenal (paragangliomas)– 10-15% are prevertebral and paravertebral sympathetic ganglia of the chest, abdomen, and pelvis.
Classification based on biochemical secretory pattern
Pheochromocytoma and paragangliomas (PPGL) can be classified based on the biochemical secretory pattern: [2]
- Noradrenergic phenotype (predominant norepinephrine secreting)- associated with Von Hippel-Lindau syndrome
- Adrenergic phenotype (predominant epinephrine secreting)- associated with MEN2 or neurofibromatosis type 1 (NF1)
- Dopamine secreting- associated with SDHB, SDHD or SDHC mutations and potentially metastatic tumors.
Classification based on genetics
Familial pheochromocytoma
- Familial pheochromocytoma is associated with several hereditary disorders such as:
- Multiple Endocrine Neoplasia types 2A
- 2B (MEN2) (caused by mutations of the RET gene)
- Von Hippel-Lindau (VHL) disease (caused by mutations of the VHL gene)
- Familial paraganglioma of the neck (cause by mutations of the gene for succinate dehydrogenase subunit D (SDHD))
- Neurofibromatosis type 1 (NF1)
Non-familial pheochromocytoma
- Resulting from sporadic germ-line mutations, which have been documented in about 20% of cases.
- The majority of them are positive for KIT expression. A partial explanation was provided by the finding of activating mutations in another gene encoding an RTK, the platelet-derived growth factor receptor alpha (PDGFRA) gene in some KIT-negative GISTs
Sporadic
- Most catecholamine-secreting tumors are sporadic. Mutations have been identified in most of the sporadic cases.
- Sporadic tumors may be due to spontaneous mutation, decreased penetrance or maternal imprinting.[5]
- 50% of patients had a pathogenic mutation in SDHB, SDHD, or VHL.[6]
Classification based on mutations and pathogenetic pathways
Pheochromocytoma and paragangliomas (PPGL) can be classified into the following clusters- [7] [8] [9]
- Cluster 1
- Mutations involving in overexpression of vascular endothelial growth factor (VEGF) as a result of pseudohypoxia
- Impaired DNA methylation leading to increased vascularization
- Cluster 2
- Activating mutations of Wnt-signaling pathway including Wnt receptor signaling and Hedgehog signaling.
- Mutations of CSDE1 (Cold shock domain containing E1) and MAML3 (Mastermind like transcriptional coactivator 3) genes7.
- Cluster 3
- Abnormal activation of kinase signaling pathways like PI3Kinase/AKT, RAS/RAF/ERK, and mTOR pathways.
References
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ 2.0 2.1 Smith RJ, Bryant RG (1975). “Metal substitutions incarbonic anhydrase: a halide ion probe study”. Biochem Biophys Res Commun. 66 (4): 1281–6. doi:10.1016/0006-291x(75)90498-2. PMID orcid.org/0000-0003-2771-564X Check
|pmid=value (help). - ↑ Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P; et al. (2002). “Biochemical diagnosis of pheochromocytoma: which test is best?”. JAMA. 287 (11): 1427–34. doi:10.1001/jama.287.11.1427. PMID 11903030.
- ↑ Lenders JW, Eisenhofer G, Mannelli M, Pacak K (2005). “Phaeochromocytoma”. Lancet. 366 (9486): 665–75. doi:10.1016/S0140-6736(05)67139-5. PMID 16112304.
- ↑ Buffet A, Venisse A, Nau V, Roncellin I, Boccio V, Le Pottier N; et al. (2012). “A decade (2001-2010) of genetic testing for pheochromocytoma and paraganglioma”. Horm Metab Res. 44 (5): 359–66. doi:10.1055/s-0032-1304594. PMID 22517557.
- ↑ Jafri M, Whitworth J, Rattenberry E, Vialard L, Kilby G, Kumar AV; et al. (2013). “Evaluation of SDHB, SDHD and VHL gene susceptibility testing in the assessment of individuals with non-syndromic phaeochromocytoma, paraganglioma and head and neck paraganglioma”. Clin Endocrinol (Oxf). 78 (6): 898–906. doi:10.1111/cen.12074. PMID 23072324.
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Eisenhofer G, Huynh TT, Pacak K, Brouwers FM, Walther MM, Linehan WM; et al. (2004). “Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome”. Endocr Relat Cancer. 11 (4): 897–911. doi:10.1677/erc.1.00838. PMID 15613462.
- ↑ Lam AK (2017). “Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours”. Endocr Pathol. 28 (3): 213–227. doi:10.1007/s12022-017-9484-5. PMID 28477311.
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
It is understood that pheochromocytoma is mediated by excessive secretion of catecholamines and subsequent stimulation of adrenergic receptors. It arises from the chromaffin cells of the adrenal medulla and sympathetic ganglia. The pathophysiology of pheochromocytoma does not depend on the histological subtype. Malignant and benign pheochromocytomas share the same biochemical and histological features. It may be sporadic or familial. All of these forms have genetic origin depending on a large number of genes, for example, VHL, SDH, NF1, RET genes. It is associated with conditions like MEN 2A syndrome, MEN 2B syndrome, VHL disease, and NF1.
Pathophysiology
Physiology
Pheochromocytoma is not associated with normal physiology.
Pathology
- It is understood that pheochromocytoma is the is mediated by excessive secretion of catecholamines and subsequent stimulation of adrenergic receptors.
- Commonly secreted catecholamines include norepinephrine (predominant) and epinephrine. Some tumors may also secrete dopamine. [1]
- Excessive secretion of catecholamines may be either continuous or intermittent.
- Pheochromocytoma is a tumor which arises from the chromaffin cells of the adrenal medulla and sympathetic ganglia.
- The pathophysiology of pheochromocytoma does not depend on the histological subtype. Malignant and benign pheochromocytomas share the same biochemical and histological features. [2][3][4]
- The exact mechanism responsible for surge in catecholamine secretion remains unclear but it has been postulated that certain medications (such as opiates, metoclopramide or beta blockers) and changes in tumor blood flow and pressure could be responsible factors.
- Binding to β1 receptors causes renin release from juxtaglomerular cells and lipolysis in adipose tissue. It Increases cardiac output by:
- Increase in heart rate in sinoatrial node
- Increase in atrial cardiac muscle contractility
- Increases in contractility and automaticity of ventricular cardiac muscle
- Increases in conduction and automaticity of atrioventricular node
Genetics
- Genes involved in the pathogenesis of pheochromocytoma include:
- RET gene (MEN 2A, MEN 2B syndromes)
- NF1 gene
- VHL gene (VHL disease)
- SDHD, SDHB, and SDHC genes of the mitochondrial complex [7]
- SDHA, SDHAF2, TMEM127 (transmembrane protein 127), MAX (myc-associated factor X), FH (fumarate hydratase), PDH1, PDH2 (pyruvate dehydrogenase), HIF1alpha (hypoxia-inducible factor), MDH2 (malate dehydrogenase), and KIF1Bß (kinesin family member) genes. [8]
Pheochromocytoma and paragangliomas (PPGL) susceptibility genes can be classified into the following clusters- [9] [10] [11]
- Cluster 1
- Mutations involving in overexpression of vascular endothelial growth factor (VEGF) as a result of pseudohypoxia
- Impaired DNA methylation leading to increased vascularization
- Cluster 2
- Activating mutations of Wnt-signaling pathway including Wnt receptor signaling and Hedgehog signaling.
- Mutations of CSDE1 (Cold shock domain containing E1) and MAML3 (Mastermind like transcriptional coactivator 3) genes7.
- Cluster 3
- Abnormal activation of kinase signaling pathways like PI3Kinase/AKT, RAS/RAF/ERK, and mTOR pathways.
Associated conditions
Conditions associated with pheochromocytoma include:
- Multiple endocrine neoplasia (MEN1)
- Multiple endocrine neoplasia (MEN2B)
- Von-Hippel Lindau disease (VHL)
- Neurofibromatosis 1 (NF1)
| MEN 1 | MEN 2 |
|---|---|
Gross Pathology
On gross pathology, the characteristic findings of pheochromocytoma are:
- Small to large tumors usually associated with hemorrhage and necrosis.[12]
- Usually lobulated
- Bilateral when familial tumors
- Associated with hyperplasia in the adjacent medulla.
- Chromaffin reaction: fresh tumor cut section turns dark brown if add potassium dichromate at pH 5-6.
-
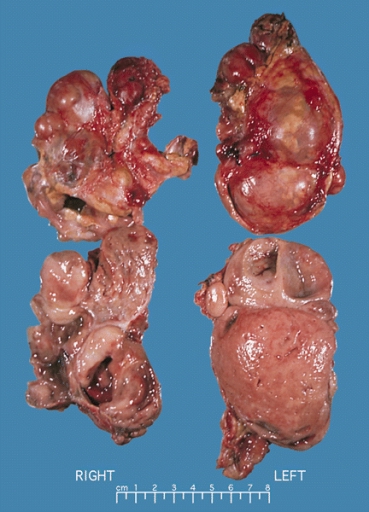 Bilateral pheochromocytoma in MEN2. Gross image. Source: https://upload.wikimedia.org/wikipedia/commons/5/5f/Bilateral_pheo_MEN2.jpg
Bilateral pheochromocytoma in MEN2. Gross image. Source: https://upload.wikimedia.org/wikipedia/commons/5/5f/Bilateral_pheo_MEN2.jpg

Microscopic Pathology
On microscopic histopathological analysis, the characterisitc findings of pheochromocytoma typically include: [13] [14]
- A nesting (Zellballen) pattern- this pattern is composed of well-defined clusters of tumor cells (round or polygonal epithelioid cells) containing eosinophilic cytoplasm separated by fibrovascular stroma.
- These cells have a central nucleus with an eosinophilic, granular cytoplasm, and clumped chromatin.
- At the periphery, spindle-shaped sustentacular or supporting cells are seen.
-
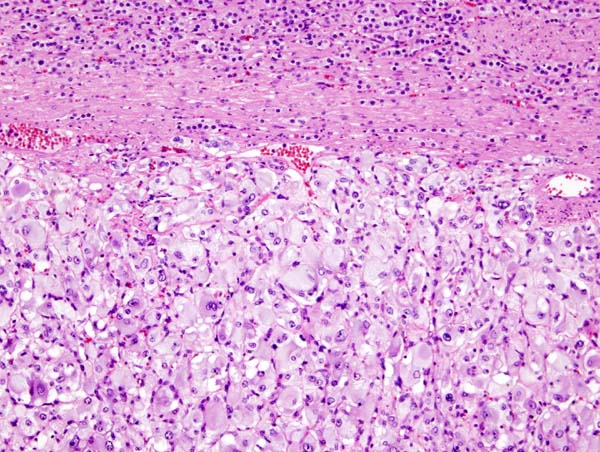 Micrograph of pheochromocytoma. Source: By Nephron – Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524
Micrograph of pheochromocytoma. Source: By Nephron – Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524 -
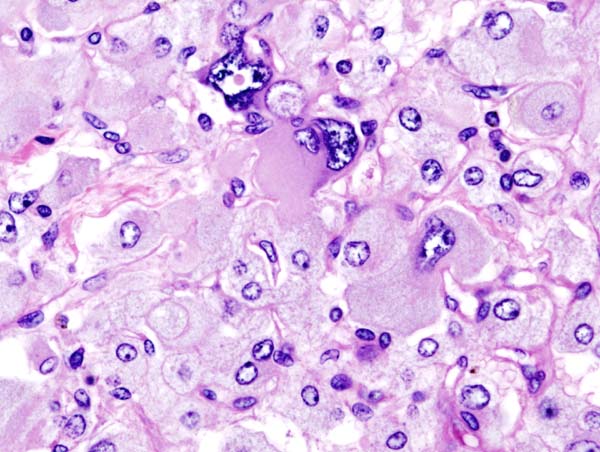 Histopathology of adrenal pheochromocytoma. Adrenectomy specimen. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535945
Histopathology of adrenal pheochromocytoma. Adrenectomy specimen. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535945 -
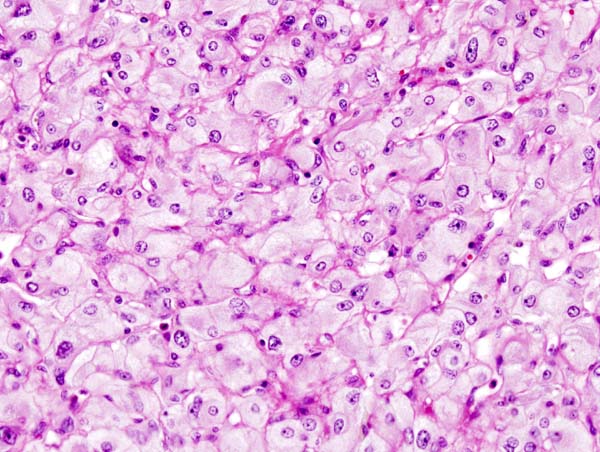 Micrograph of pheochromocytoma. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535944
Micrograph of pheochromocytoma. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535944



Videos
{{#ev:youtube|7yjxG3KmX98}}
References
- ↑ Smith RJ, Bryant RG (1975). “Metal substitutions incarbonic anhydrase: a halide ion probe study”. Biochem Biophys Res Commun. 66 (4): 1281–6. doi:10.1016/0006-291x(75)90498-2. PMID orcid.org/0000-0003-2771-564X Check
|pmid=value (help). - ↑ Goldstein RE, O’Neill JA, Holcomb GW, Morgan WM, Neblett WW, Oates JA; et al. (1999). “Clinical experience over 48 years with pheochromocytoma”. Ann Surg. 229 (6): 755–64, discussion 764-6. PMC 1420821. PMID 10363888.
- ↑ Raz I, Katz A, Spencer MK (1991). “Epinephrine inhibits insulin-mediated glycogenesis but enhances glycolysis in human skeletal muscle”. Am J Physiol. 260 (3 Pt 1): E430–5. PMID 1900669.
- ↑ Arnall DA, Marker JC, Conlee RK, Winder WW (1986). “Effect of infusing epinephrine on liver and muscle glycogenolysis during exercise in rats”. Am J Physiol. 250 (6 Pt 1): E641–9. PMID 3521311.
- ↑ Webb TA, Sheps SG, Carney JA (1980). “Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2”. Am. J. Surg. Pathol. 4 (2): 121–6. PMID 6103678.
- ↑ Yee JK, Moores JC, Jolly DJ, Wolff JA, Respess JG, Friedmann T (1987). “Gene expression from transcriptionally disabled retroviral vectors”. Proc. Natl. Acad. Sci. U.S.A. 84 (15): 5197–201. PMC 298821. PMID 3474647.
- ↑ Gimm O (2005). “Pheochromocytoma-associated syndromes: genes, proteins and functions of RET, VHL and SDHx”. Fam Cancer. 4 (1): 17–23. doi:10.1007/s10689-004-5740-1. PMID 15883706.
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Eisenhofer G, Huynh TT, Pacak K, Brouwers FM, Walther MM, Linehan WM; et al. (2004). “Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome”. Endocr Relat Cancer. 11 (4): 897–911. doi:10.1677/erc.1.00838. PMID 15613462.
- ↑ Lam AK (2017). “Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours”. Endocr Pathol. 28 (3): 213–227. doi:10.1007/s12022-017-9484-5. PMID 28477311.
- ↑ Sajjanar AB, Athanikar VS, Dinesh US, Nanjappa B, Patil PB (2015). “Non Functional Unilateral Adrenal Myelolipoma, A Case Report”. J Clin Diagn Res. 9 (6): ED03–4. doi:10.7860/JCDR/2015/13209.6070. PMC 4525519. PMID 26266130.
- ↑ Kliewer KE, Wen DR, Cancilla PA, Cochran AJ (1989). “Paragangliomas: assessment of prognosis by histologic, immunohistochemical, and ultrastructural techniques”. Hum Pathol. 20 (1): 29–39. doi:10.1016/0046-8177(89)90199-8. PMID 2912871.
- ↑ Kliewer KE, Cochran AJ (1989). “A review of the histology, ultrastructure, immunohistology, and molecular biology of extra-adrenal paragangliomas”. Arch Pathol Lab Med. 113 (11): 1209–18. PMID 2684087.
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
The most common cause of pheochromocytoma is sporadic mutations. Less common causes of pheochromocytoma include familial associations and association with syndromes. Familial pheochromocytoma may be caused by a mutation of either SDHD, VHL, SDHB, RET, NF1 genes.
Causes
Life-threatening Causes
- Pheochromocytoma due to any cause may be life-threatening which may result in death.
Common Causes
- In most cases of pheochromocytoma, the cause is unknown.
- Sporadic form is more common
Less Common Causes
Less common causes of pheochromocytoma include:
- Familial form
- Associated with syndromes- Neurofibromatosis 1, Von Hippel-Lindau disease, Multiple Endocrine Neoplasia 2A and 2B
Genetic Causes
Pheochromocytoma of the familial type may be caused by a mutation in the following genes:
- RET gene (MEN 2A, MEN 2B syndromes)
- NF1 gene
- VHL gene (VHL disease)
- SDHD, SDHB, and SDHC genes of the mitochondrial complex [1]
- SDHA, SDHAF2, TMEM127 (transmembrane protein 127), MAX (myc-associated factor X), FH (fumarate hydratase), PDH1, PDH2 (pyruvate dehydrogenase), HIF1alpha (hypoxia-inducible factor), MDH2 (malate dehydrogenase), and KIF1Bß (kinesin family member) genes. [2]
Pheochromocytoma and paragangliomas (PPGL) susceptibility genes can be classified into the following clusters- [3] [4] [5]
- Cluster 1
- Mutations involving in overexpression of vascular endothelial growth factor (VEGF) as a result of pseudohypoxia
- Impaired DNA methylation leading to increased vascularization
- Cluster 2
- Activating mutations of Wnt-signaling pathway including Wnt receptor signaling and Hedgehog signaling.
- Mutations of CSDE1 (Cold shock domain containing E1) and MAML3 (Mastermind like transcriptional coactivator 3) genes7.
- Cluster 3
- Abnormal activation of kinase signaling pathways like PI3Kinase/AKT, RAS/RAF/ERK, and mTOR pathways.
Causes by Organ System
| Cardiovascular | No underlying causes |
| Chemical/Poisoning | No underlying causes |
| Dental | No underlying causes |
| Dermatologic | No underlying causes |
| Drug Side Effect | No underlying causes |
| Ear Nose Throat | No underlying causes |
| Endocrine | No underlying causes |
| Environmental | No underlying causes |
| Gastroenterologic | No underlying causes |
| Genetic | No underlying causes |
| Hematologic | No underlying causes |
| Iatrogenic | No underlying causes |
| Infectious Disease | No underlying causes |
| Musculoskeletal/Orthopedic | No underlying causes |
| Neurologic | No underlying causes |
| Nutritional/Metabolic | No underlying causes |
| Obstetric/Gynecologic | No underlying causes |
| Oncologic | No underlying causes |
| Ophthalmologic | No underlying causes |
| Overdose/Toxicity | No underlying causes |
| Psychiatric | No underlying causes |
| Pulmonary | No underlying causes |
| Renal/Electrolyte | No underlying causes |
| Rheumatology/Immunology/Allergy | No underlying causes |
| Sexual | No underlying causes |
| Trauma | No underlying causes |
| Urologic | No underlying causes |
| Miscellaneous | No underlying causes |
Causes in Alphabetical Order
List of causes of the disease in alphabetical order:
- Familial inheritance
- FH (fumarate hydratase) gene
- HIF1alpha (hypoxia-inducible factor) gene
- KIF1Bß (kinesin family member) gene
- MAX (myc-associated factor X)
- MDH2 (malate dehydrogenase) gene
- NF1 gene– Neurofibromatosis 1 (NF1)
- PDH1, PDH2 (pyruvate dehydrogenase) gene
- RET gene- Multiple endocrine neoplasia (MEN 2A, MEN 2B)
- SDHA gene
- SDHB gene
- SDHC gene
- SDHD gene
- SDHAF2 gene
- Sporadic mutations
- TMEM127 gene
- Unknown origin
- VHL gene– Von-Hippel Lindau disease (VHL)
References
- ↑ Gimm O (2005). “Pheochromocytoma-associated syndromes: genes, proteins and functions of RET, VHL and SDHx”. Fam Cancer. 4 (1): 17–23. doi:10.1007/s10689-004-5740-1. PMID 15883706.
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Eisenhofer G, Huynh TT, Pacak K, Brouwers FM, Walther MM, Linehan WM; et al. (2004). “Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome”. Endocr Relat Cancer. 11 (4): 897–911. doi:10.1677/erc.1.00838. PMID 15613462.
- ↑ Lam AK (2017). “Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours”. Endocr Pathol. 28 (3): 213–227. doi:10.1007/s12022-017-9484-5. PMID 28477311.
Differentiating Pheochromocytoma from other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
Pheochromocytoma must be differentiated from other causes of paroxysmal hypertension including severe paroxysmal hypertension (pseudopheochromocytoma), panic disorder, factitious hypertension, carcinoid syndrome, migraine headache, hyperthyroidism, renovascular hypertension, hypoglycemia, labile hypertension (White coat hypertension), stroke, compression of the lateral medulla, seizures, baroreflex failure and drugs.
Differentiating pheochromocytoma from other diseases
Pheochromocytoma must be differentiated from other causes of paroxysmal hypertension. The differentials include:
| Disease | Symptoms | Signs | Investigations |
|---|---|---|---|
| Pheochromocytoma[1][4] | Features of sympathetic nervous systemhyperactivity and include:
|
|
|
| Pseudopheochromocytoma (idiopathic)[1][2][3][4] | Paroxysmal activation of the sympathetic system may cause:
|
| |
| Panic attacks |
|
|
Laboratory studies that can exclude medical disorders other than panic disorder include: |
| Labile hypertension (White coat hypertension) |
|
Elevated blood pressure, tachycardia, and may be anxiety in a clinical setting but not in other settings[1] |
|
| Hyperthyroidism |
|
|
|
| Renovascular hypertension |
|
| |
| Stroke and compression of lateral medulla (Lateral medullary syndrome) |
|
|
|
| Seizures | According to type; it may be focal or generalized, clinical or subclinical:[7]
|
|
|
| Carcinoid syndrome | Hypertensive crisis occurs with malignant carcinoid syndrome[10]. Symptoms include:
|
| |
| Migraine headaches |
|
|
CT is indicated in patients with:[1][2]
CT is not indicated in:
|
| Drugs | Sympathomimetic drugs that can induce symptoms simulating pheochromocytoma include:
|
|
|
| Baroreflex failure[18] |
|
|
|
Pheochromocytoma must be differentiated from other adrenal tumors such as adrenocortical adenoma, adrenal metastasis, and Cushing’s syndrome.
| Differential Diagnosis | Clinical picture | Imagings | Laboratory tests |
|---|---|---|---|
| Adrenocortical carcinoma |
|
|
|
| Adrenal adenoma |
|
|
|
| Cushing’s syndrome |
|
|
|
| Pheochromocytoma |
|
|
|
| Adrenal metastasis |
|
|
| S.No. | Disease | Symptoms | Signs | Diagnosis | Comments | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Abdominal Pain | Hematuria | Headache | Abdominal mass | Abdominal tenderness | Ultrasonography | CT scan | Histology | |||
| 1. | Wilms tumor | + | + | – | + | + |
|
|
|
|
| 2. | Renal cell carcinoma | + | + | +/- | + | – |
|
Both CT and MRI may be used to detect neoplastic masses that may define renal cell carcinoma or metastasis of the primary cancer. CT scan and use of intravenous (IV) contrast is generally used for work-up and follow-up of patients with renal cell carcinoma. | The histological pattern of renal cell carcinoma depends whether it is papillary, chromophobe or collecting duct renal cell carcinoma. | |
| 3. | Rhabdoid kidney disease | + | + | – | + | – |
|
|
|
|
| 4. | Polycystic kidney disease | + | + | + (from hypertension) | + | – |
Ultrasound may be helpful in the diagnosis of polycystic kidney disease. Findings on an ultrasound diagnostic of polycystic kidney disease include:[25][26] |
Renal CT scan may be helpful in the diagnosis of polycystic kidney disease. Findings on CT scan diagnostic of ADPKD include:
|
||
| 5. | Pheochromocytoma | – | – | + (as a part of the hypertension paroxysm) | – | – |
|
The following findings may be observed on CT scan:[31]
|
|
|
| 6. | Burkitt lymphoma | +/- (in non-endemic or sporadic form of the disease) | – | – | – | – |
|
|
|
|
| 7. | Intussusception | + | – | – | +/- | + |
|
|
|
|
| 8. | Hydronephrosis | + | +/- | – | – | + (CVA tenderness in case of pyelonephritis) |
|
|
|
|
| 9. | Dysplastic kidney | N/A | N/A | N/A | N/A | N/A |
MCDK is usually diagnosed by ultrasound examination before birth.
|
|
||
| 10. | Pediatric Neuroblastoma | + | – | – | +/- | +/- |
|
|
|
|
| 11. | Pediatric Rhabdomyosarcoma | + | +/- | +/- | – | +/- | On CT scan, rhabdomyosarocma is characterized by:
|
|
||
| 12. | Mesoblastic nephroma | + | + | – | + | – |
|
|
Classic mesoblastic nephroma
Cellular mesoblastic nephroma
Mixed mesoblastic nephroma
|
Most common renal tumor that occurs in 1st month of life |
References
- ↑ Mann SJ (1999). “Severe paroxysmal hypertension (pseudopheochromocytoma): understanding the cause and treatment”. Arch Intern Med. 159 (7): 670–4. PMID 10218745.
- ↑ Mann SJ (1999). “Severe paroxysmal hypertension (pseudopheochromocytoma): understanding the cause and treatment”. Arch Intern Med. 159 (7): 670–4. PMID 10218745.
- ↑ Mann SJ (1996). “Severe paroxysmal hypertension. An automatic syndrome and its relationship to repressed emotions”. Psychosomatics. 37 (5): 444–50. doi:10.1016/S0033-3182(96)71532-3. PMID 8824124.
- ↑ Sharabi Y, Goldstein DS, Bentho O, Saleem A, Pechnik S, Geraci MF; et al. (2007). “Sympathoadrenal function in patients with paroxysmal hypertension: pseudopheochromocytoma”. J Hypertens. 25 (11): 2286–95. doi:10.1097/HJH.0b013e3282ef5fac. PMID 17921824.
- ↑ Iglesias P, Acosta M, Sánchez R, Fernández-Reyes MJ, Mon C, Díez JJ (2005). “Ambulatory blood pressure monitoring in patients with hyperthyroidism before and after control of thyroid function”. Clin Endocrinol (Oxf). 63 (1): 66–72. doi:10.1111/j.1365-2265.2005.02301.x. PMID 15963064.
- ↑ Mintz G, Pizzarello R, Klein I (1991). “Enhanced left ventricular diastolic function in hyperthyroidism: noninvasive assessment and response to treatment”. J Clin Endocrinol Metab. 73 (1): 146–50. doi:10.1210/jcem-73-1-146. PMID 2045465.
- ↑ 7.0 7.1 Mintz G, Pizzarello R, Klein I (1991). “Enhanced left ventricular diastolic function in hyperthyroidism: noninvasive assessment and response to treatment”. J Clin Endocrinol Metab. 73 (1): 146–50. doi:10.1210/jcem-73-1-146. PMID 2045465.
- ↑ Brigo F, Storti M, Lochner P, Tezzon F, Fiaschi A, Bongiovanni LG; et al. (2012). “Tongue biting in epileptic seizures and psychogenic events: an evidence-based perspective”. Epilepsy Behav. 25 (2): 251–5. doi:10.1016/j.yebeh.2012.06.020. PMID 23041172.
- ↑ Fountain NB, Van Ness PC, Swain-Eng R, Tonn S, Bever CT, American Academy of Neurology Epilepsy Measure Development Panel and the American Medical Association-Convened Physician Consortium for Performance Improvement Independent Measure Development Process (2011). “Quality improvement in neurology: AAN epilepsy quality measures: Report of the Quality Measurement and Reporting Subcommittee of the American Academy of Neurology”. Neurology. 76 (1): 94–9. doi:10.1212/WNL.0b013e318203e9d1. PMID 21205698.
- ↑ Warner RR, Mani S, Profeta J, Grunstein E (1994). “Octreotide treatment of carcinoid hypertensive crisis”. Mt Sinai J Med. 61 (4): 349–55. PMID 7969229.
- ↑ Sjöblom SM (1988). “Clinical presentation and prognosis of gastrointestinal carcinoid tumours”. Scand J Gastroenterol. 23 (7): 779–87. PMID 3227292.
- ↑ Feldman JM (1986). “Urinary serotonin in the diagnosis of carcinoid tumors”. Clin Chem. 32 (5): 840–4. PMID 2421946.
- ↑ Eriksson B, Arnberg H, Oberg K, Hellman U, Lundqvist G, Wernstedt C; et al. (1990). “A polyclonal antiserum against chromogranin A and B–a new sensitive marker for neuroendocrine tumours”. Acta Endocrinol (Copenh). 122 (2): 145–55. PMID 2316306.
- ↑ Sundin A, Vullierme MP, Kaltsas G, Plöckinger U, Mallorca Consensus Conference participants. European Neuroendocrine Tumor Society (2009). “ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: radiological examinations”. Neuroendocrinology. 90 (2): 167–83. doi:10.1159/000184855. PMID 19077417.
- ↑ Kelman L (2004). “The premonitory symptoms (prodrome): a tertiary care study of 893 migraineurs”. Headache. 44 (9): 865–72. doi:10.1111/j.1526-4610.2004.04168.x. PMID 15447695.
- ↑ Krentz AJ, Mikhail S, Cantrell P, Hill GM (2001). “Drug Points: Pseudophaeochromocytoma syndrome associated with clozapine”. BMJ. 322 (7296): 1213. PMC 31620. PMID 11358774.
- ↑ Kuchel O (1985). “Pseudopheochromocytoma”. Hypertension. 7 (1): 151–8. PMID 3980057.
- ↑ Robertson D, Hollister AS, Biaggioni I, Netterville JL, Mosqueda-Garcia R, Robertson RM (1993). “The diagnosis and treatment of baroreflex failure”. N Engl J Med. 329 (20): 1449–55. doi:10.1056/NEJM199311113292003. PMID 8413455.
- ↑ 19.0 19.1 Zar T, Peixoto AJ (2008). “Paroxysmal hypertension due to baroreflex failure”. Kidney Int. 74 (1): 126–31. doi:10.1038/ki.2008.30. PMID 18322544.
- ↑ Manolopoulou J, Fischer E, Dietz A, Diederich S, Holmes D, Junnila R; et al. (2015). “Clinical validation for the aldosterone-to-renin ratio and aldosterone suppression testing using simultaneous fully automated chemiluminescence immunoassays”. J Hypertens. 33 (12): 2500–11. doi:10.1097/HJH.0000000000000727. PMID 26372319.
- ↑ Hartman DS, Sanders RC (April 1982). “Wilms’ tumor versus neuroblastoma: usefulness of ultrasound in differentiation”. J Ultrasound Med. 1 (3): 117–22. PMID 6152936.
- ↑ De Campo JF (1986). “Ultrasound of Wilms’ tumor”. Pediatr Radiol. 16 (1): 21–4. PMID 3003660.
- ↑ Cahan LD (1985). “Failure of encephalo-duro-arterio-synangiosis procedure in moyamoya disease”. Pediatr Neurosci. 12 (1): 58–62. PMID 4080660.
- ↑ Jolly RD, Stellwagen E, Babul J, Vodkaĭlo LV, Titov VL, Moldomusaev DM, Maianskiĭ AN (November 1975). “Mannosidosis of Angus Cattle: a prototype control program for some genetic diseases”. Adv Vet Sci Comp Med. 19 (23): 1–21. PMID 1978.
- ↑ Chapman AB, Devuyst O, Eckardt KU, Gansevoort RT, Harris T, Horie S, Kasiske BL, Odland D, Pei Y, Perrone RD, Pirson Y, Schrier RW, Torra R, Torres VE, Watnick T, Wheeler DC (July 2015). “Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference”. Kidney Int. 88 (1): 17–27. doi:10.1038/ki.2015.59. PMC 4913350. PMID 25786098.
- ↑ Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, San Millan JL, Gibson R, Breuning M, Peters D, Ravine D (January 2009). “Unified criteria for ultrasonographic diagnosis of ADPKD”. J. Am. Soc. Nephrol. 20 (1): 205–12. doi:10.1681/ASN.2008050507. PMC 2615723. PMID 18945943.
- ↑ Stavrou C, Koptides M, Tombazos C, Psara E, Patsias C, Zouvani I, Kyriacou K, Hildebrandt F, Christofides T, Pierides A, Deltas CC (October 2002). “Autosomal-dominant medullary cystic kidney disease type 1: clinical and molecular findings in six large Cypriot families”. Kidney Int. 62 (4): 1385–94. doi:10.1111/j.1523-1755.2002.kid581.x. PMID 12234310.
- ↑ Bleyer AJ, Kmoch S, Antignac C, Robins V, Kidd K, Kelsoe JR, Hladik G, Klemmer P, Knohl SJ, Scheinman SJ, Vo N, Santi A, Harris A, Canaday O, Weller N, Hulick PJ, Vogel K, Rahbari-Oskoui FF, Tuazon J, Deltas C, Somers D, Megarbane A, Kimmel PL, Sperati CJ, Orr-Urtreger A, Ben-Shachar S, Waugh DA, McGinn S, Bleyer AJ, Hodanová K, Vylet’al P, Živná M, Hart TC, Hart PS (March 2014). “Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1”. Clin J Am Soc Nephrol. 9 (3): 527–35. doi:10.2215/CJN.06380613. PMC 3944763. PMID 24509297.
- ↑ Faguer S, Decramer S, Chassaing N, Bellanné-Chantelot C, Calvas P, Beaufils S, Bessenay L, Lengelé JP, Dahan K, Ronco P, Devuyst O, Chauveau D (October 2011). “Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood”. Kidney Int. 80 (7): 768–76. doi:10.1038/ki.2011.225. PMID 21775974.
- ↑ Heidet L, Decramer S, Pawtowski A, Morinière V, Bandin F, Knebelmann B, Lebre AS, Faguer S, Guigonis V, Antignac C, Salomon R (June 2010). “Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases”. Clin J Am Soc Nephrol. 5 (6): 1079–90. doi:10.2215/CJN.06810909. PMC 2879303. PMID 20378641.
- ↑ Bravo EL (1991). “Pheochromocytoma: new concepts and future trends”. Kidney Int. 40 (3): 544–56. PMID 1787652.
- ↑ Whalen RK, Althausen AF, Daniels GH (1992). “Extra-adrenal pheochromocytoma”. J Urol. 147 (1): 1–10. PMID 1729490.
- ↑ Baid SK, Lai EW, Wesley RA, Ling A, Timmers HJ, Adams KT; et al. (2009). “Brief communication: radiographic contrast infusion and catecholamine release in patients with pheochromocytoma”. Ann Intern Med. 150 (1): 27–32. PMC 3490128. PMID 19124817.
- ↑ Bravo EL (1991). “Pheochromocytoma: new concepts and future trends”. Kidney Int. 40 (3): 544–56. PMID 1787652.
- ↑ Burkitt lymphoma. MedlinePlus. https://www.nlm.nih.gov/medlineplus/ency/article/001308.htm Accessed on September 30, 2015
- ↑ Bellan C, Lazzi S, De Falco G, Nyongo A, Giordano A, Leoncini L (2003). “Burkitt’s lymphoma: new insights into molecular pathogenesis”. J. Clin. Pathol. 56 (3): 188–92. PMC 1769902. PMID 12610094. Unknown parameter
|month=ignored (help) - ↑ Ko HS, Schenk JP, Tröger J, Rohrschneider WK (2007). “Current radiological management of intussusception in children”. Eur Radiol. 17 (9): 2411–21. doi:10.1007/s00330-007-0589-y. PMID 17308922.
- ↑ Boyle MJ, Arkell LJ, Williams JT (1993). “Ultrasonic diagnosis of adult intussusception”. Am. J. Gastroenterol. 88 (4): 617–8. PMID 8470658.
- ↑ Neuroblastoma. Radiopaedia (2015) http://radiopaedia.org/articles/neuroblastoma Accessed on October, 8 2015
- ↑ Colon NC, Chung DH (2011). “Neuroblastoma”. Adv Pediatr. 58 (1): 297–311. doi:10.1016/j.yapd.2011.03.011. PMC 3668791. PMID 21736987.
- ↑ Neuroblastoma. Radiopaedia (2015) http://radiopaedia.org/articles/neuroblastoma Accessed on October, 8 2015
- ↑ Neuroblastoma. Libre Pathology(2015) http://librepathology.org/wiki/index.php/Adrenal_gland#Neuroblastoma Accessed on October, 5 2015
- ↑ Mesoblastic nephroma.Dr Ayush Goel and Dr Yuranga Weerakkody et al. Radiopaedia.org 2015. http://radiopaedia.org/articles/mesoblastic-nephroma
Epidemiology and Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2]Mohammed Abdelwahed M.D[3]
Overview
The incidence of pheochromocytoma ranges 0.2-0.8 per 100,000 persons. The median age at diagnosis is 24.9 years in familial cases and 43.9 years in sporadic cases. Both men and women are affected equally by pheochromocytoma.
Epidemiology and Demographics
Incidence
- In the USA, the incidence of pheochromocytoma ranges from 0.2-0.8 per 100,000 persons.[1] [2]
- Annually reported cases range from 500 to 1600 in the United States.[3]
- Autopsy studies have discovered a higher number of cases than the actual prevalence rates. Ten percent of pheochromocytomas cases are discovered by chance.
Prevalence
- In the USA, the prevelance of pheochromocytoma ranges from 0.2-0.8 per 100,000 persons.[1]
- The prevalence of pheochromocytoma in patients with hypertension in general outpatient clinics ia about 0.1%. [4]
- The prevalence of pheochromocytoma is approximately 1.7% in children with hypertension.
- About 5% of patients with incidentally discovered adrenal masses on imaging actually have pheochromocytoma.
- The prevalence of pheochromocytoma in individuals carrying a germline mutation in pheochromocytoma susceptibility genes may be around 50%.
Case-fatality rate/Mortality rate
- The incidence of pheochromocytoma ranges from 0.2-0.8 per 100,000 persons with a perioperative case-fatality rate/mortality rate of about 2.4%.[1] [5]
- Metastatic PPGLs were associated with a 2.40-fold higher risk of mortality than non-metastatic PPGLs (95% confidence interval, 1.38 to 4.17; P=0.002). [6]
Age
- Patients of all age groups may develop pheochromocytoma. Approximately 10% occur in children.
- The median age at diagnosis is 40 years.
- The average age at diagnosis is 24.9 years in hereditary cases and 43.9 years in sporadic cases.[1]
- Hereditary tumors present at a younger age than sporadic.
Race
- There is no racial predilection to pheochromocytoma. [7]
Gender
- Pheochromocytoma affects men and women are equally. [1]
Region
- Pheochromocytoma is a rare disease that tends to affect all populations equally.
Developed Countries
Developing Countries
References
- ↑ 1.0 1.1 1.2 1.3 1.4 National Cancer Institute. Physician Data Query Database 2015. National Cancer Institute. Physician Data Query Database 2015. http://www.cancer.gov/types/pheochromocytoma/hp/pheochromocytoma-treatment-pdq#link/_25_toc
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K; et al. (2010). “The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer”. Pancreas. 39 (6): 775–83. doi:10.1097/MPA.0b013e3181ebb4f0. PMC 3419007. PMID 20664475.
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Plouin PF, Duclos JM, Soppelsa F, Boublil G, Chatellier G (2001). “Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center”. J Clin Endocrinol Metab. 86 (4): 1480–6. doi:10.1210/jcem.86.4.7392. PMID 11297571.
- ↑ Kim JH, Moon H, Noh J, Lee J, Kim SG (2020). “Epidemiology and Prognosis of Pheochromocytoma/Paraganglioma in Korea: A Nationwide Study Based on the National Health Insurance Service”. Endocrinol Metab (Seoul). 35 (1): 157–164. doi:10.3803/EnM.2020.35.1.157. PMC 7090309 Check
|pmc=value (help). PMID 32207276 Check|pmid=value (help). - ↑ DE GRAEFF J, HORAK BJ (1964). “THE INCIDENCE OF PHAEOCHROMOCYTOMA IN THE NETHERLANDS”. Acta Med Scand. 176: 583–93. doi:10.1111/j.0954-6820.1964.tb00661.x. PMID 14223534.
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Mohammed Abdelwahed M.D[2] Ifrah Fatima, M.B.B.S[3]
Overview
The most potent risk factor for pheochromocytoma is a family history of multiple endocrine neoplasias, Von Hippel-Lindau disease, neurofibromatosis type 1, hereditary paraganglioma syndromes.
Risk Factors
The most potent risk factor in the development of pheochromocytoma is a family history of multiple endocrine neoplasias, Von Hippel-Lindau disease, neurofibromatosis type 1 or hereditary paraganglioma syndromes.
Common Risk Factors
- Common risk factors in the development of pheochromocytoma include harboring the following genes:
Less Common Risk Factors
- Less common risk factors in the development of pheochromocytoma include harboring the following genes:
References
- ↑ Gimm O (2005). “Pheochromocytoma-associated syndromes: genes, proteins and functions of RET, VHL and SDHx”. Fam Cancer. 4 (1): 17–23. doi:10.1007/s10689-004-5740-1. PMID 15883706.
- ↑ Jameson, J (2017). Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
Screening
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Mohammed Abdelwahed M.D[2]
Overview
Familial pheochromocytoma is associated with multiple endocrine neoplasias, VHL and neurofibromatosis1 and should be screened by plasma fractionated metanephrines levels. The next step is to obtain 24-hour urinary fractionated metanephrine levels. Imaging should be considered if the initial tests are positive. Genetic testing also should be performed in high-risk patients.
Screening
Biochemical screening
- According to the Endocrine Society, biochemical screening for pheochromocytoma in recommended among patients with:
- VHL syndrome– started at 5 years of age with biochemical surveillance every year for the rest of life.
- Signs or symptoms suggesting catecholamine excess, especially if the symptoms are paroxysmal.
- Unexpected blood pressure changes to drugs, surgery, or anesthesia
- Unexplained blood pressure variability
- Incidentaloma, even if the patient is normotensive
- Blood pressure that is difficult to control
- History of previous treatment for pheochromocytoma or paraganglioma
- Hereditary risk of pheochromocytoma or paraganglioma in family members
- Syndromic features relating to a pheochromocytoma-related hereditary syndromes [1]
- Plasma fractionated metanephrine level is the best test. If elevated, 24-hour urinary fractionated metanephrines should be done.
Imaging screening
Anatomic imaging should be used when norepinephrine levels are elevated more than two times upper normal limits.[2]
- For high-risk children, screening for pheochromocytoma should begin by 11 years of age.
- For moderate risk patients, screening should be started by 16 years of age.
- If positive, adrenal imaging (CT) or (MRI) should be performed.
Genetic screening
- Genetic testing should be performed in:[1]
- Patients with a family history of pheochromocytoma
- Tumors or malignant or extra-adrenal pheochromocytoma
- Families whose infants or young children have Hirschsprung disease
- First-degree relatives of a patient with proven germline RET mutation
- Patients with cutaneous lichen amyloidosis
- Patients with known RET mutations.
- Parents whose young children have MEN 2A/2B
References
- ↑ 1.0 1.1 Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH; et al. (2014). “Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline”. J Clin Endocrinol Metab. 99 (6): 1915–42. doi:10.1210/jc.2014-1498. PMID 24893135.
- ↑ Aufforth RD, Ramakant P, Sadowski SM, Mehta A, Trebska-McGowan K, Nilubol N; et al. (2015). “Pheochromocytoma Screening Initiation and Frequency in von Hippel-Lindau Syndrome”. J Clin Endocrinol Metab. 100 (12): 4498–504. doi:10.1210/jc.2015-3045. PMC 4667160. PMID 26451910.
Natural History, Complications and Prognosis
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
Pheochromocytoma is an adrenaline secreting tumor, that usually develops in the fifth decade of life. Symptoms start with tachycardia, hypertension, headache and sweating. Massive release of catecholamines may cause hyperglycemia, malignant hypertension and metastasis. The prognosis of pheochromocytoma is generally good but metastatic pheochromocytoma has a 5-year survival rate of approximately 50%.
Natural History, Complications and Prognosis
Natural History
- The symptoms of pheochromocytoma usually develop in the fifth decade of life.
- Common symptoms are with tachycardia, hypertension, headache, and sweating.
- If left untreated, hyperglycemia and hypertensive emergency. It may lead to heart failure andcerebrovascular strokes.
- If malignant, It can metastasize to lymph nodes, bones, lungs, and liver. [1]
Complications
Common complications of pheochromocytoma include:
- Damage to cardiac myocytes due to either a compromise of the coronary microcirculation or the direct toxic effects of catecholamines on cardiac myocytes.[2]
- Hyperglycemia due to opposition of insulin effect by high doses of adrenaline secreted by the tumor.
- Malignant hypertension that may cause cerebrovascular accidents such as:
- Metastasis to:
Prognosis
- The prognosis of pheochromocytoma is generally good with treatment. The 5-year survival rate in patients with metastatic pheochromocytoma is approximately 50%.[3]
- Prognosis and survival rate varies with the location of the primary tumor, sites of metastases, Tumor burden, and rate of progression.
- Metastasis to the brain and liver has a worse prognosis than other metastases.
Post-surgical prognosis
- Factors associated with a favourable prognosis include:
- Small tumor size
- Short duration of surgery
- Systolic blood pressure < 160 mmHg
- Low levels of urinary catecholamines.[4]
- Approximately 10% recur after being resected.
- Recurrence is more common in patients with familial pheochromocytoma and extra-adrenal tumors.[5]
References
- ↑ Pamporaki C, Hamplova B, Peitzsch M, Prejbisz A, Beuschlein F, Timmers HJLM; et al. (2017). “Characteristics of Pediatric vs Adult Pheochromocytomas and Paragangliomas”. J Clin Endocrinol Metab. 102 (4): 1122–1132. doi:10.1210/jc.2016-3829. PMID 28324046.
- ↑ Goldman 2011, pp. 327
- ↑ National Cancer Institute. Physician Data Query Database 2015. http://www.cancer.gov/types/pheochromocytoma/hp/pheochromocytoma-treatment-pdq#link/_25_toc
- ↑ Murphy MM, Witkowski ER, Ng SC, McDade TP, Hill JS, Larkin AC; et al. (2010). “Trends in adrenalectomy: a recent national review”. Surg Endosc. 24 (10): 2518–26. doi:10.1007/s00464-010-0996-z. PMID 20336320.
- ↑ Plouin PF, Duclos JM, Soppelsa F, Boublil G, Chatellier G (2001). “Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center”. J Clin Endocrinol Metab. 86 (4): 1480–6. doi:10.1210/jcem.86.4.7392. PMID 11297571.
Diagnosis
Diagnosis
History and Symptoms | Physical Examination | Laboratory Findings | Electrocardiogram | X Ray | CT | MRI | Echocardiography or Ultrasound | Other Imaging Findings | Other Diagnostic Studies
Treatment
Treatment
Medical Therapy | Surgery | Primary Prevention | Secondary Prevention | Cost-Effectiveness of Therapy | Future or Investigational Therapies
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH