
Cystic fibrosis
For patient information click here
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Shaghayegh Habibi, M.D.[2]
Synonyms and keywords: CF; mucoviscoidosis; mucoviscidosis
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Alberto Castro Molina, M.D.
Overview
Cystic fibrosis is an autosomal recessive disease that caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The genetic mutation result in defective transport of chloride, and secondarily sodium and eventually abnormal viscous mucoid secretions mostly in lungs and GI tract. Infertility due to atresia/absent vasa deferentia and abnormal/absent seminal vesicles is the associated condition of cystic fibrosis. Cystic fibrosis has to be differentiated from asthma, bronchiolitis, emphysema and primary ciliary dyskinesia (Kartagener syndrome). Immunoreactive trypsinogen (IRT) of serum is raised in newborns with cystic fibrosis and has been used as a screening test. The most significant complications are seen in airways and most common chronic pulmonary infection include P. aeruginosa, S. aureus and H. influenzae. Gastrointestinal complications include pancreatic insufficiency, pancreatitis, gastroesophageal reflux disease, distal intestinal obstruction syndrome, constipation and small intestinal bacterial overgrowth. The sweat chloride test is the gold standard test for the diagnosis of cystic fibrosis. Most common symptoms include salty sweat, constant coughing, diarrhea or greasy stools, stomach pain, constipation and poor weight gain. Also abdominal distension and digital clubbing may be detected. Lung examination may presents hyperresonant lungs, Wheeze or crackles. Most common chest CT scan findings include peribronchial thickening, mucous plugging and Bronchiectasis. Medical treatments has targeted following consequences of the defect such as GI and pulmonary mucus plugging and infection. Treatment include mucolytic agents (dornase alfa, N-acetyl-L-cysteine), airway surface rehydration, anti-infective agents, anti-inflammatory agents and potentiators of CFTR protein defect.
Over the past decade, CFTR modulator therapies have transformed the natural history of cystic fibrosis for many patients, with improvements in lung function, pulmonary exacerbations, nutritional status, and quality of life, particularly with highly effective triple therapy (elexacaftor/tezacaftor/ivacaftor) in people with at least one Phe508del allele.[1] As a result, the cystic fibrosis population is increasingly adult, with evolving care needs and a growing focus on comorbidities, treatment burden, and long-term outcomes.[2][3]
Historical Perspective
In the late 1930s cystic fibrosis was first recognized as a disease. In 1949, Lowe and colleagues suggested this theory that cystic fibrosis must be caused by a defect in a single gene. In 1959, the measurement of sweat electrolyte concentrations was established as the mainstay of diagnosing CF. In 1989, the CFTR gene was discovered first. In 1990, scientists successfully added cloned normal gene to cystic fibrosis cells which corrected the chloride transportion.
The era of disease-modifying therapy began with CFTR modulators, including ivacaftor (a CFTR potentiator) for gating mutations such as G551D, followed by combination modulator regimens for Phe508del (including lumacaftor/ivacaftor and tezacaftor/ivacaftor) and highly effective triple therapy (elexacaftor/tezacaftor/ivacaftor).[4][5][6][7][1]
Classification
Cystic fibrosis may be classified according to CFTR protein function abnormality into 6 groups: lack of production (Class 1), failure to reach the site of action due to misfolding (class 2), defects in gating (class 3), reduced ion conductance (class 4), abnormally low channel numbers (class 5) and decreased half-life (class 6). Cystic fibrosis classes 1,2 and 3 are the most common types which have associated with pancreatic insufficiency.
Eligibility for CFTR modulators depends on CFTR genotype and functional consequences of the variant(s). Curated resources (e.g., CFTR2) support genotype interpretation and guide therapy selection in clinical practice and newborn screening follow-up.[8][9]
Pathophysiology
Cystic fibrosis is an autosomal recessive disease that caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Substitution of a single amino acid is the most common type of CFTR gene mutation. CFTR gene functions as a chloride channel (pumps chloride from the intracellular space to the extracellular space) found on the surface of the epithelial cells. The genetic mutations result in defective transport of chloride, and secondarily sodium and eventually abnormal viscous mucoid secretions mostly in lungs (results in airway surface liquid depletion, decreased mucociliary transport, inflammation and infection) and GI tract (results in reduced volume of pancreatic secretion, pancreatic tissue destruction and fibrosis, malnutrition and poor growth). Infertility due to atresia/absent vasa deferentia and abnormal/absent seminal vesicles is the associated condition of cystic fibrosis.
Experimental and translational studies support airway surface liquid depletion and impaired mucociliary clearance as early drivers of cystic fibrosis lung disease, including evidence from large-animal (porcine) models that demonstrate reduced airway surface liquid and impaired bacterial clearance associated with CFTR dysfunction.[10] Reviews of early lung disease and pathogenesis emphasize that airway obstruction, infection, and inflammation begin early in life and contribute to progressive structural lung injury.[11]
Causes
Cystic fibrosis is caused by mutations in the CFTR gene. The genetic mutations result in defective transport of chloride, and secondarily sodium, by epithelial cells and eventually abnormal viscous mucoid secretions mostly in lungs and GI tract.
Differentiating cystic fibrosis from Other Diseases
Cystic fibrosis has to be differentiated from other conditions with similar presentation of cough and wheeze like common cold, asthma, bronchiolitis, emphysema, primary ciliary dyskinesia (Kartagener syndrome), bronchitis, bronchiectasis, foreign body aspiration, pneumoconiosis, interstitial lung disease, cardiogenic pulmonary edema, GERD and sarcoidosis.
Epidemiology and Demographics
The incidence of cystic fibrosis is approximately 1 in 2500 livebirths. It is a life-limiting disease (100% mortality rate), and a cure for the disease remains elusive. Most patients with cystic fibrosis are diagnosed in first 2 years of life. The onset of symptoms is before the first month of life in 12%, between 1-6 months of age in 75%, and between 6-12 months of age in 7% of patients. Although cystic fibrosis has been reported in all racial and ethnic groups, it mostly affects Caucasians of Northern European descent. It affects men and women equally.
Contemporary registry and population-based analyses demonstrate improved survival and an increasing proportion of adults living with cystic fibrosis, with differences by region and health system. Cystic fibrosis population trends and survival estimates are frequently monitored through national registries and comparative cohort analyses.[3][12][2]
Risk Factors
Every person inherits two CFTR genes, one from each parent. Children who inherit two mutated CFTR genes from both parents will have cystic fibrosis.
Screening
Newborn screening identified most of the children with cystic fibrosis before the symptoms develop. It offers this opportunity for early diagnosis and improved outcomes. Immunoreactive trypsinogen (IRT) of serum is raised in newborns with cystic fibrosis and has been used as a screening test. A raised IRT in the first week of life is a sensitive test but not specific for cystic fibrosis.
In many screening programs, elevated IRT is followed by CFTR variant testing, and in some algorithms, a sequencing tier is used to improve diagnostic yield and reduce false positives and inequities across ancestry groups. Screening approaches vary by region and program design.[13]
Natural History, Complications, and Prognosis
Malnutrition and poor growth (due to loss of pancreatic exocrine function) leads to death in the first decade of life for most untreated patients. The most significant complications are seen in airways (responsible for 80% of mortality) and most common chronic pulmonary infection include P. aeruginosa, S. aureus and H. influenzae. In cystic fibrosis 98% of men are infertile due to aspermia. Lung complications are currently the primary causes of morbidity and are responsible for 80% of mortality in these patients and gastrointestinal complications include pancreatic insufficiency, pancreatitis, gastroesophageal reflux disease, distal intestinal obstruction syndrome, constipation and small intestinal bacterial overgrowth. In cystic fibrosis, obstructive lung disease and other lung complications are currently the primary causes of morbidity and are responsible for 80% of mortality. At present time survival probability of children is 40-50 years. Women with cystic fibrosis have a shortened life expectancy compared to men.
As CFTR modulators have become widely used, outcomes and complications are changing, including improvements in nutritional status and lung function in many treated individuals. Emerging clinical questions include how to safely reduce treatment burden in selected patients receiving highly effective CFTR modulators.[14][2]
Diagnosis
Diagnostic study of choice
The sweat chloride test is the gold standard test for the diagnosis of cystic fibrosis. A sweat chloride value of more than 59 mmol/L is diagnostic for cystic fibrosis, 30-59 mmol/L needs more evaluation with CFTR genetic analysis and less than 30 is indicates that cystic fibrosis is unlikely.
Cystic fibrosis diagnostic criteria and guidance for ambiguous or intermediate sweat chloride results incorporate CFTR genotyping, functional testing when needed, and careful clinical correlation. Expert guidelines also address CFTR-related disorders and diagnostic uncertainty.[15][8]
History and Symptoms
Most common symptoms in cystic fibrosis include salty sweat, constant coughing, diarrhea or greasy stools, stomach pain, constipation and poor weight gain. Less common symptoms include nasal polyp, hemoptysis and skin irritation.
Physical Examination
In cystic fibrosis abdominal distension and digital clubbing may be detected. In HEENT examination there is nasal polyps and signs of rhinosinusitis (purulent nasal discharge, mucosal edema, turbinate hypertrophy and tenderness on palpitation of the sinuses). Lung examination may presents hyperresonant lungs, Wheeze or crackles and Productive cough with mucoid or purulent sputum.
Laboratory Findings
Immunoreactive trypsinogen (IRT) of serum is raised in newborns with cystic fibrosis and has been used as a screening test.
Electrocardiogram
There are no electrocardiogram findings associated with cystic fibrosis.
X-ray
In cystic fibrosis the chest radiographic features may overlap with many other disorders, particularly those characterized by inflammatory or destructive changes of the airways. Atelectasis is common in infancy. Most patients with CF demonstrate some of the classic chest radiographic findings that reflect chronic bronchiectasis include hyperinflation, bronchial thickening and dilatation, peribronchial cuffing, mucoid impaction, cystic radiolucencies, increase in interstitial marking and cattered nodular densities.
Ultrasound
In cystic fibrosis, ultrasound findings include small cystic degeneration could be observed in the pancreatic tail. Transabdominal ultrasound of the pancreas demonstrated a higher pancreatic echogenicity, as a measure of pancreatic lipomatosis in pancreatic insufficient CF patients. Echogenic bowel is found on ultrasound in 50% to 78% of fetuses affected with cystic fibrosis. It is thought to be caused by changes in the consistency of meconium in the small intestine as a result of abnormalities in pancreatic enzyme secretion. The sonographic findings include diffuse echogenic bowel, focal echogenic bowel with calcifications, hyperechoic mass and bowel dilation.
CT scan
Computed tomography (CT scan) findings in patients with cystic fibrosis are more sensitive as compared to the pulmonary function tests. Most common chest CT scan findings include peribronchial thickening, mucous plugging and Bronchiectasis. Less common findings include abscesses, emphysematous bullae, hyperinflation, collapse, consolidation, ground-glass opacities, acinar nodules and thickening of interlobular and intralobular septa. Abdomen CT scan in patients with cystic fibrosis may include these findings diffuse and complete fatty replacement of pancreas, Fibrosis of the pancreas and Intestinal obstruction.
MRI
MRI may be helpful in determining the cause of linear lung markings in cystic fibrosis. It is also helpful in differentiating mucous plugging and peribronchial thickening from normal pulmonary blood vessels. Because of MRI absence of ionising radiation and possibility for obtaining functional information, it is helpful for assessing lung disease in children who require repetitive follow up imaging for a long time. MRI studies of the pancreas have demonstrated different patterns of fatty infiltration, ductal changes, pancreatic cysts, calcifications and hypoechoic areas representing fibrosis.
Other Imaging Findings
There are no other imaging findings for cystic fibrosis.
Other Diagnostic Studies
Other diagnostic studies in patients with cystic fibrosis include sweat chloride test (measures the chloride content of the sweat) and nasal potential differences (performed by running different solutions through the nose) which used to detect changes in CFTR function. A sweat chloride value of more than 59 mmol/L is diagnostic for cystic fibrosis and less than 30 mmol/L indicates that cystic fibrosis is unlikely. Also Pulmonary function test (PFT) is important in monitoring lung function in patients with cystic fibrosis. However, it is only an indirect measure of lung structure and is insensitive to local or early damage.
Treatment
Medical Therapy
Medical treatments for patients with cystic fibrosis has targeted following consequences of the defect such as GI and pulmonary mucus plugging and infection. Treatment include mucolytic agents (dornase alfa, N-acetyl-L-cysteine), airway surface rehydration (hypertonic saline, osmotic agents), anti-infective agents (for prophylaxis, eradication of early infection and suppression of chronic infection), anti-inflammatory agents (NSAIDs, inhaled corticosteroids, LTB4 receptor antagonists and Azithromycin) and potentiators of CFTR protein defect.
CFTR modulator therapy
CFTR modulators are genotype-directed therapies that include potentiators (e.g., ivacaftor) and correctors (e.g., lumacaftor, tezacaftor, elexacaftor) used alone or in combination. These therapies improve CFTR function and have been associated with improved lung function, reduced pulmonary exacerbations, and improved nutritional parameters in eligible patients.[9]
- Ivacaftor for gating mutations such as G551D was associated with clinically meaningful improvements in outcomes in pivotal trials.[4]
- Combination corrector-potentiator therapies for Phe508del (including lumacaftor/ivacaftor and tezacaftor/ivacaftor) expanded modulator eligibility and provided more modest benefits than later triple therapy.[5][6][7]
- Elexacaftor/tezacaftor/ivacaftor (highly effective triple therapy) provides substantial improvements in patients with at least one Phe508del allele and is expected to influence long-term outcomes across respiratory and extrapulmonary domains.[1]
As modulator use increases, clinical trials and observational data are evaluating how to safely reduce treatment burden (e.g., discontinuation of hypertonic saline or dornase alfa) in selected patients receiving highly effective modulators.[14]
Airway clearance and mucolytics
Airway clearance therapies remain foundational for many patients and are supported by guideline-based recommendations.[16] Dornase alfa and hypertonic saline have evidence of benefit in selected populations and are often used to reduce mucus plugging and exacerbations.[17][18]
Chronic airway infection and inhaled antibiotics
Chronic airway infection and inflammation are major drivers of morbidity. Guidance supports early eradication and chronic suppressive therapy for Pseudomonas aeruginosa infection, and inhaled antibiotics are a cornerstone of management.[19][20] Opportunistic pathogens (including nontuberculous mycobacteria and fungi) can complicate management and may require specialized evaluation and therapy.[21]
Extrapulmonary and comorbidity management
CFTR modulators may alter extrapulmonary manifestations, including reports of improved pancreatic function in selected individuals treated with ivacaftor.[22] CF-related diabetes and other metabolic complications remain important in adult care; registry analyses provide updated estimates of prevalence and trends.[23] Mental health screening and management are increasingly emphasized, including clinician guidance and observational studies evaluating symptom changes after initiation of highly effective modulators.[24][25]
Emerging therapies
For patients not eligible for current modulators or with persistent disease burden, investigational approaches include antisense oligonucleotide therapies for specific splicing mutations, respiratory gene therapy, and inhaled mRNA-based therapies.[26][27][28]
Surgery
Cystic fibrosis patients with a large pneumothorax should undergo chest tube insertion and even surgical pleurodesis in case of recurrent large pneumothorax. When medical treatment for pulmonary complications fails, lung transplantation is the only option.
Lung transplantation remains an important option for advanced lung disease, and contemporary registry studies describe outcomes in cystic fibrosis recipients.[29]
Primary Prevention
There is no known way for the primary prevention of cystic fibrosis.
Secondary Prevention
In cystic fibrosis secondary prevention include airway clearance techniques, dornase alpha, hypertonic saline, antibiotics, immunizations, physical activity, nutritional support for pancreatic insufficiency and extra salt and water.
References
- ↑ 1.0 1.1 1.2 Middleton PG, Mall MA, Dřevínek P; et al. (2019). “Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele”. N Engl J Med. 381: 1809–1819.
- ↑ 2.0 2.1 2.2 Burgel PR, Burnet E, Regard L, Martin C. (2023). “The changing epidemiology of cystic fibrosis: the implications for adult care”. Chest. 163: 89–99.
- ↑ 3.0 3.1 “Cystic Fibrosis Foundation patient registry 2021 annual data report” (PDF). Cystic Fibrosis Foundation. 2022.
- ↑ 4.0 4.1 Ramsey BW, Davies J, McElvaney NG; et al. (2011). “A CFTR potentiator in patients with cystic fibrosis and the G551D mutation”. N Engl J Med. 365: 1663–1672.
- ↑ 5.0 5.1 Wainwright CE, Elborn JS, Ramsey BW; et al. (2015). “Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR”. N Engl J Med. 373: 220–231.
- ↑ 6.0 6.1 Taylor-Cousar JL, Munck A, McKone EF; et al. (2017). “Tezacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del”. N Engl J Med. 377: 2013–2023.
- ↑ 7.0 7.1 Rowe SM, Daines C, Ringshausen FC; et al. (2017). “Tezacaftor–ivacaftor in residual-function heterozygotes with cystic fibrosis”. N Engl J Med. 377: 2024–2035.
- ↑ 8.0 8.1 “The Clinical and Functional TRanslation of CFTR (CFTR2)”. 2011.
- ↑ 9.0 9.1 Despotes KA, Donaldson SH. (2022). “Current state of CFTR modulators for treatment of cystic fibrosis”. Curr Opin Pharmacol. 65: 102239.
- ↑ Pezzulo AA, Tang XX, Hoegger MJ; et al. (2012). “Reduced airway surface liquid and defective bacterial clearance in the porcine cystic fibrosis lung”. Nature. 487: 109–113.
- ↑ Stoltz DA, Meyerholz DK, Welsh MJ. (2015). “Origins of cystic fibrosis lung disease”. N Engl J Med. 372: 351–362.
- ↑ Stephenson AL, Swaleh S, Sykes J; et al. (2023). “Contemporary survival comparison in cystic fibrosis in Canada and the United States”. J Cyst Fibros. 22: 443–449.
- ↑ Currier RJ, Sciortino S, Liu R, Bishop T, Alikhani Koupaei R; et al. (2017). “Genetic testing in cystic fibrosis newborn screening: is IRT plus two variants followed by third-tier sequencing?”. Genet Med. 19: 1159–1163.
- ↑ 14.0 14.1 Mayer-Hamblett N, Ratjen F, Russell R; et al. (2023). “Discontinuation of hypertonic saline or dornase alfa in modulator-treated people with cystic fibrosis: a platform of non-inferiority trials”. Lancet Respir Med. 11: 329–340.
- ↑ Farrell PM, White TB, Ren CL; et al. (2017). “Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation”. J Pediatr. 181: S4–S15.e1.
- ↑ Flume PA, Robinson KA, O’Sullivan BP; et al. (2009). “Cystic fibrosis pulmonary guidelines: airway clearance therapies”. Respir Care. 54: 522–537.
- ↑ Yang C, Montgomery M. (2021). “Dornase alfa for cystic fibrosis”. Cochrane Database Syst Rev. 3: CD001127.
- ↑ Elkins MR, Robinson M, Rose BR; et al. (2006). “A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis”. N Engl J Med. 354: 229–240.
- ↑ Mogayzel PJ Jr, Naureckas ET, Robinson KA; et al. (2014). “Cystic fibrosis pulmonary guidelines. Pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection”. Ann Am Thorac Soc. 11: 1640–1650.
- ↑ Smith S, Rowbotham NJ. (2022). “Inhaled anti-pseudomonal antibiotics for long-term therapy in cystic fibrosis”. Cochrane Database Syst Rev. 11: CD001021.
- ↑ Blanchard AC, Waters VJ. (2022). “Opportunistic pathogens in cystic fibrosis: epidemiology and pathogenesis of lung infection”. J Pediatric Infect Dis Soc. 11: Suppl 2:S3-S12.
- ↑ Hutchinson I, McNally P. (2021). “Appearance of pancreatic sufficiency after discontinuation of pancreatic enzyme replacement therapy in children with cystic fibrosis on ivacaftor”. Ann Am Thorac Soc. 18: 182–183.
- ↑ Szentpetery S, Fernandez GS, Schechter MS, Jain R, Flume PA; et al. (2022). “Cystic fibrosis-related diabetes: prevalence, trends and associated factors data from the US Cystic Fibrosis Foundation patient registry”. J Cyst Fibros. 21: 777–783.
- ↑ “Depression, anxiety, and cystic fibrosis: guide for CF clinicians” (PDF). Cystic Fibrosis Foundation. 2021.
- ↑ Zhang L, Albon D, Jones M, Bruschwein H. (2022). “Impact of elexacaftor/tezacaftor/ivacaftor on depression and anxiety in cystic fibrosis”. Ther Adv Respir Dis. 16: 17534666221144211.
- ↑ Oren YS, Irony-Tur Sinai M, Golec A; et al. (2021). “Antisense oligonucleotide therapy in cystic fibrosis patients carrying the 3849+10 kb C-to- to-T splicing mutation”. J Cyst Fibros. 20: 865–875.
- ↑ McLachlan G, Alton EWFW, Boyd AC; et al. (2022). “Progress in respiratory gene therapy”. Hum Gene Ther. 33: 893–912.
- ↑ Rowe SM, Zuckerman JB, Dorgan D; et al. (2023). “Inhaled mRNA therapy for cystic fibrosis: interim results of a randomized, double-blind, placebo-controlled phase 1/2 clinical study”. J Cyst Fibros. 22: 656–664.
- ↑ Yeung JC, Machuca TN, Chaparro C; et al. (2020). “Lung transplantation outcomes for cystic fibrosis”. J Heart Lung Transplant. 39: 553–560.
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
In the late 1930s, cystic fibrosis was first recognized as a disease. In 1949, Lowe and colleagues suggested this theory that cystic fibrosis must be caused by a defect in a single gene. In 1959, the measurement of sweat electrolyte concentrations was established as the mainstay of diagnosing CF. In 1989, the CFTR gene was discovered first. In 1990, scientists successfully added cloned normal gene to cystic fibrosis cells which corrected the chloride transportion.
Historical Perspective
Discovery
- In the late 1930s, cystic fibrosis was first recognized as a disease. This term was used to describe the characteristic cyst formation and fibrosis observed in the pancreas.[1][2][3]
- In 1949, Lowe and colleagues suggested that cystic fibrosis must be caused by a defect in a single gene, based on autosomal recessive pattern of inheritance.
- In 1959, the measurement of sweat electrolyte concentrations was established as the mainstay of diagnosing CF.
- In the late 1980s, the first articles described the application of computed tomography in patients with cystic fibrosis.
- In 1989, the CFTR gene was discovered.
- Till 1996, approximately 500 different mutations for cystic fibrosis had been detected.[4]
Landmark Events in the Development of Treatment Strategies
- In 1990, scientists successfully added cloned normal gene to cystic fibrosis cells in the laboratory, which corrected the chloride transportion. The gene therapy technique was then tried on a limited number of CF patients.[5]
References
- ↑ Pettit RS, Fellner C (July 2014). “CFTR Modulators for the Treatment of Cystic Fibrosis”. P T. 39 (7): 500–11. PMC 4103577. PMID 25083129.
- ↑ Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW (August 2008). “Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report”. J. Pediatr. 153 (2): S4–S14. doi:10.1016/j.jpeds.2008.05.005. PMC 2810958. PMID 18639722.
- ↑ Rybacka A, Karmelita-Katulska K (2016). “The Role of Computed Tomography in Monitoring Patients with Cystic Fibrosis”. Pol J Radiol. 81: 141–5. doi:10.12659/PJR.896051. PMC 4821342. PMID 27103945.
- ↑ Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW (August 2008). “Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report”. J. Pediatr. 153 (2): S4–S14. doi:10.1016/j.jpeds.2008.05.005. PMC 2810958. PMID 18639722.
- ↑ National Center for Biotechnology Information (US). Genes and Disease [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 1998-. Cystic fibrosis. [Updated 2011 Jan 31]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22202/
Classification
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
Cystic fibrosis may be classified according to CFTR protein function abnormality into 6 classes and includes lack of production (class 1), failure to reach the site of action due to misfolding (class 2), defects in gating (class 3), reduced ion conductance (class 4), abnormally low channel numbers (class 5), and decreased half-life (class 6). Cystic fibrosis classes 1,2 and 3 are the most commonly associated with pancreatic insufficiency.
Classification
Cystic fibrosis may be classified according to CFTR mutation type:[1][2][3]
| Cystic fibrosis classification according to CFTR protein function abnormality | ||
|---|---|---|
| Class | Type of abnormality | Features |
| Class 1 | Total or partial lack of production of a functional CFTR |
|
| Class 2 | Failure to reach the site of action on the cell surface
(due to misfolding of the protein) | |
| Class 3 | Defects in gating
(fail to open in response to intracellular signal) | |
| Class 4 | Reduced ion conductance |
|
| Class 5 | Abnormally low channel numbers
(splicing mutations resulting in reduced amounts of CFTR protein) | |
| Class 6 | Decreased half-life | |
References
- ↑ Burney TJ, Davies JC (2012). “Gene therapy for the treatment of cystic fibrosis”. Appl Clin Genet. 5: 29–36. doi:10.2147/TACG.S8873. PMC 3681190. PMID 23776378.
- ↑ Ratjen F, Döring G (2003). “Cystic fibrosis”. Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185.
- ↑ Edmondson C, Davies JC (2016). “Current and future treatment options for cystic fibrosis lung disease: latest evidence and clinical implications”. Ther Adv Chronic Dis. 7 (3): 170–83. doi:10.1177/2040622316641352. PMC 4907071. PMID 27347364.
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
Cystic fibrosis is an autosomal recessive disease that caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Substitution of a single amino acid is the most common type of CFTR gene mutation. CFTR gene functions as a chloride channel (pumps chloride from the intracellular space to the extracellular space) found on the surface of the epithelial cells. The genetic mutations result in defective transport of chloride, and secondarily sodium and eventually abnormal viscous mucoid secretions mostly in lungs (results in airway surface liquid depletion, decreased mucociliary transport, inflammation and infection) and GI tract (results in reduced volume of pancreatic secretion, pancreatic tissue destruction and fibrosis, malnutrition and poor growth). Infertility due to atresia/absent vasa deferentia and abnormal/absent seminal vesicles is the associated condition of cystic fibrosis.
Pathophysiology
Pathogenesis
- Cystic fibrosis (CF) is an autosomal recessive disease that caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.[1][2][3]
- CFTR gene functions as a chloride channel (pumps chloride from the intracellular space to the extracellular space) found on the surface of the epithelial cells, that line multiple organs especially lungs and GI tract.
- The genetic mutations result in defective transport of chloride, and secondarily sodium, by epithelial cells, and eventually abnormal viscous mucoid secretions mostly in lungs and GI tract.
- Other organs containing epithelia such as the sweat glands, biliary duct, the male reproductive tract, and the intestine are also affected.
- Two mechanisms which cause airway-surface-liquid depletion are as follow:
| Lack of CFTR normal activity | |||||||||||||||||||||
| Less chloride secretion | More sodium absorption | ||||||||||||||||||||
| Less water transport into the epithelial surface layer | Excessive sodium and water absorption through the epithelial channel | ||||||||||||||||||||
Lung involvement in cystic fibrosis
- In patients with cystic fibrosis abnormal chloride conductance of epithelial cells results in airway surface liquid depletion and decreased mucociliary transport (airway surface liquid is essential to support ciliary function).
- The consequence of cystic fibrosis is a vicious circle of inflammation, tissue damage and infection.[3]
- Also, exaggerated, generalized, and prolonged inflammatory response of lungs to bacterial and viral pathogen is observed.
- The inflammatory response is characterized by neutrophilic dominated airway inflammation which is present even in clinically stable patients and in young infants diagnosed by neonatal screening.[3][4]
- Breakdown of accumulated neutrophils in the infected lungs of patients with cystic fibrosis leads to the release of large amounts of DNA.
- Accumulated DNA causes high viscosity of the infected sputum, followed by decreased ciliary transport and function.[5]
Gastrointestinal tract involvement in cystic fibrosis
Pancreatic disease:
- In cystic fibrosis, approximately 90% of patients present with exocrine pancreatic insufficiency.
- Pancreatic disease results from a reduced volume of pancreatic secretion with low concentrations of bicarbonate, followed by retained and prematurely activated digestive proenzymes in pancreatic ducts, resulting in tissue destruction and fibrosis.[6]
- Abnormally viscous secretions in the ducts of the pancreas, followed by loss of pancreatic exocrine function results in malnutrition and poor growth.[4]
Biliary disease:
- One third of patients with cystic fibrosis have abnormal results on liver function tests.
- Fatty infiltration is reported in up to 70% of older patients and in nearly 10% of these progresses to biliary cirrhosis.
- Histological evaluation shows duct dilatation and intraluminal concretions.
- Bile-duct epithelium becomes hyperplastic with periductal inflammation and fibrosis.
- Up to one third of patients with cystic fibrosis, a small and poorly functioning gallbladder is detected.[6]
Genetics
Cystic fibrosis is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. This gene codes for a chloride transporter regulated by cyclic AMP (cAMP)-dependent phosphorylation. There are almost 2,000 variants of CFTR gene mutations:[4]
- Substitution of a single amino acid (40%)
- Alter RNA processing including nonsense, frameshift and missplicing (36%)
- Large rearrangements of CFTR (3%)
- Promoter regions (1%)
- Neutral variants (14%)
- The effect of the remaining 6% is unclear.
- Children who inherit one mutated CFTR gene and one normal CFTR gene are “CF carriers“. CF carriers usually have no symptoms of cystic fibrosis but they can pass the mutated CFTR gene to their children.[7]

In Caucasian populations, the frequency of mutations is as follows:[9]Template:Entête tableau charte alignement
! Mutation
! Frequency
worldwide
|—–
| ΔF508
| 66.0%
|-
| G542X
| 2.4%
|—–
| G551D
| 1.6%
|-
| N1303K
| 1.3%
|—–
| W1282X
| 1.2%
|}
Associated Conditions
- In cystic fibrosis, 98% of men are infertile. The causes of aspermia include:[10]
- Atresia or absent vasa deferentia
- Abnormal or absent seminal vesicles
Gross Pathology
-
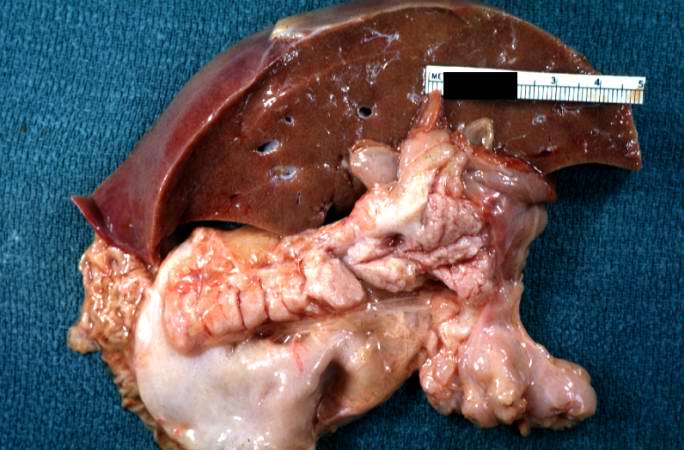 A gross photograph of liver and pancreas from the autopsy. The pancreas is slightly smaller than normal and it has a mucous consistency.
A gross photograph of liver and pancreas from the autopsy. The pancreas is slightly smaller than normal and it has a mucous consistency. -
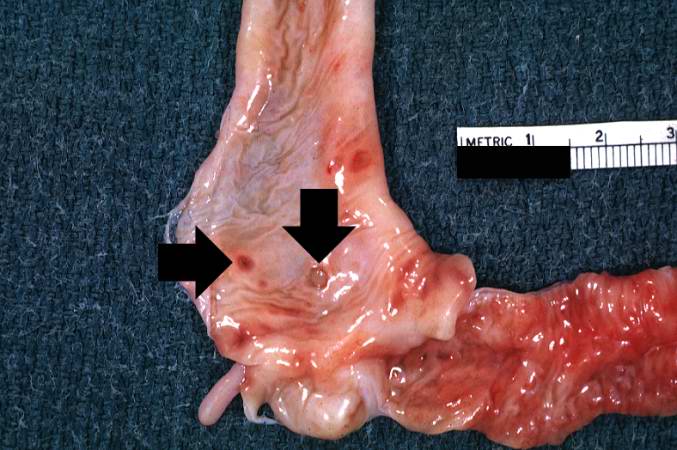 This section of duodenum demonstrates dilation, loss of rugae, and areas of ulceration (arrows).
This section of duodenum demonstrates dilation, loss of rugae, and areas of ulceration (arrows).


Microscopic Pathology
-
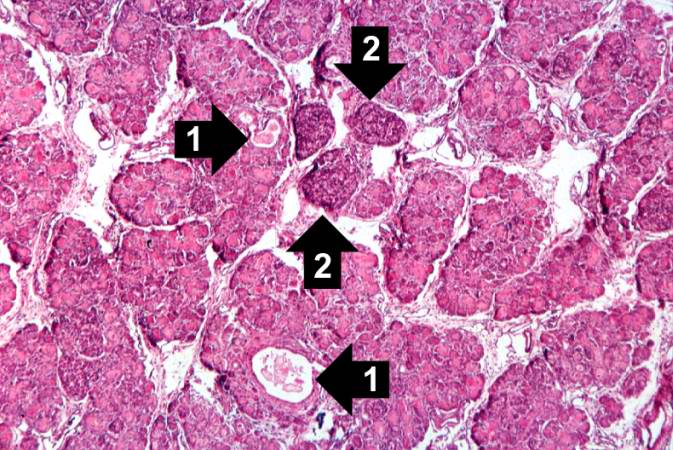 This higher-power photomicrograph of the pancreas shows interstitial tissue and the presence of small cystic spaces (1) within the acinar lobules. These spaces are filled with an eosinophilic proteinaceous material. The islets of Langerhans (2) are unaffected.
This higher-power photomicrograph of the pancreas shows interstitial tissue and the presence of small cystic spaces (1) within the acinar lobules. These spaces are filled with an eosinophilic proteinaceous material. The islets of Langerhans (2) are unaffected. -
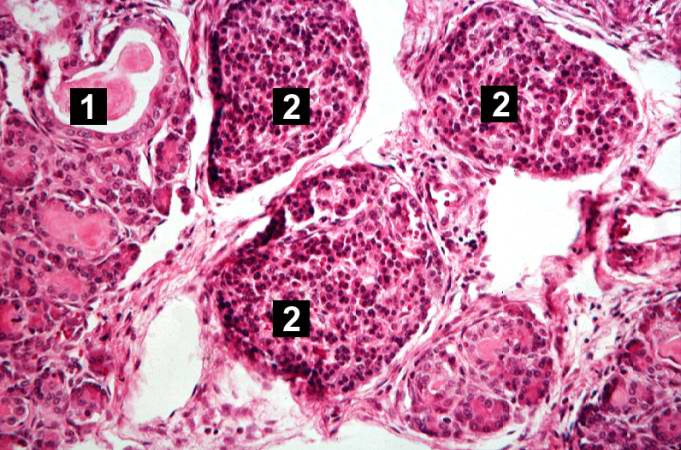 This higher-power photomicrograph shows a cystic space (1) within an acinar lobule. Islets of Langerhans (2) are also visible.
This higher-power photomicrograph shows a cystic space (1) within an acinar lobule. Islets of Langerhans (2) are also visible. -
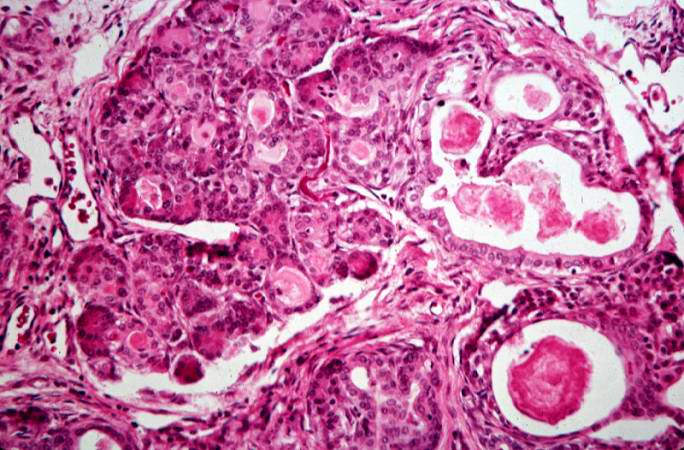 This high-power photomicrograph shows more clearly these variably-sized cystic spaces within the acinar pancreas.
This high-power photomicrograph shows more clearly these variably-sized cystic spaces within the acinar pancreas.



-
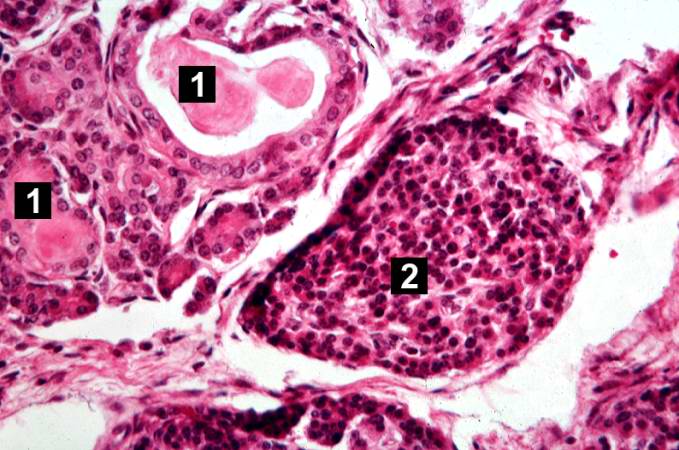 This is another high-power photomicrograph showing cystic spaces (1) within the acinar pancreas and a normal islet of Langerhans (2).
This is another high-power photomicrograph showing cystic spaces (1) within the acinar pancreas and a normal islet of Langerhans (2). -
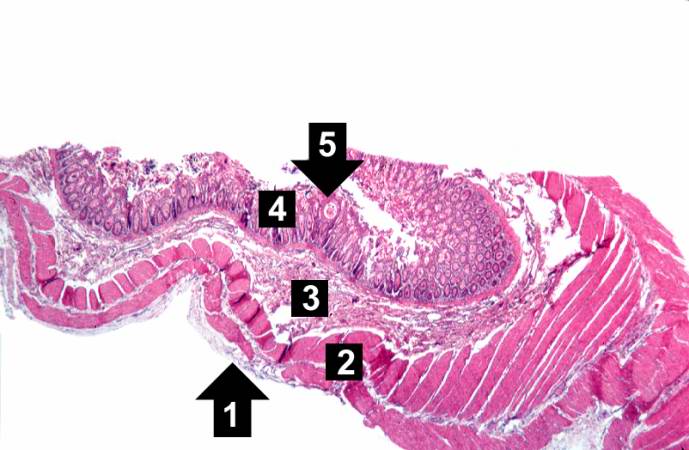 This low-power photomicrograph of intestine shows the normal layers of the intestine, including the serosa (1), the muscularis (2), the submucosa (3), and the mucosal layer (4) with its deep mucosal crypts. There is yet another cystic space within the mucosa (5).
This low-power photomicrograph of intestine shows the normal layers of the intestine, including the serosa (1), the muscularis (2), the submucosa (3), and the mucosal layer (4) with its deep mucosal crypts. There is yet another cystic space within the mucosa (5). -
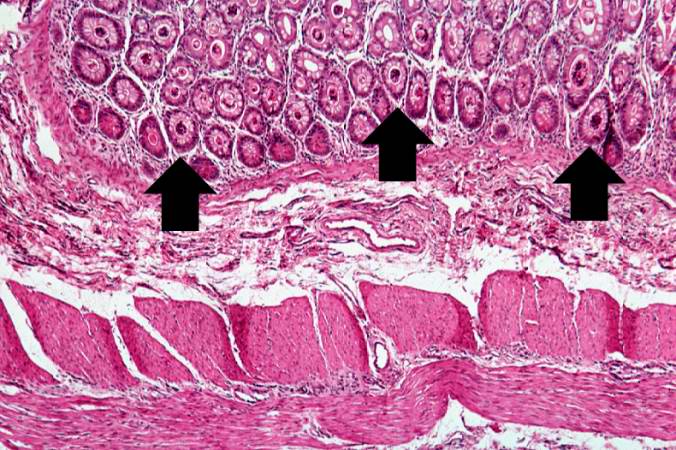 A higher-power photomicrograph shows the bottom of the intestinal crypts and the other normal layers of the intestine. Even at this magnification, accumulations of eosinophilic debris can be seen in many of the intestinal crypts (arrows).
A higher-power photomicrograph shows the bottom of the intestinal crypts and the other normal layers of the intestine. Even at this magnification, accumulations of eosinophilic debris can be seen in many of the intestinal crypts (arrows).



-
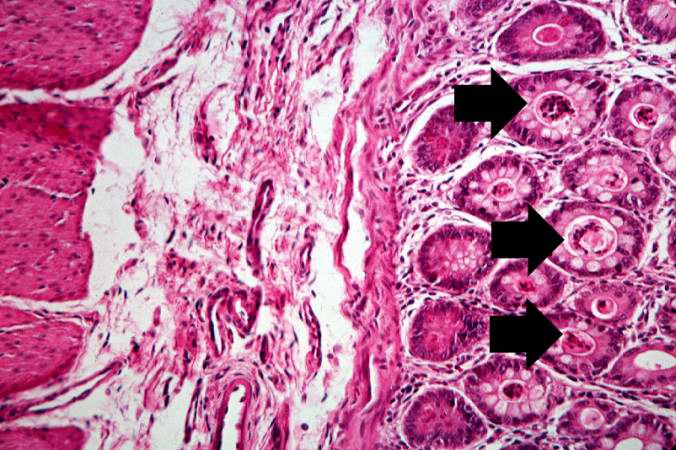 This is a higher-power photomicrograph showing the eosinophilic debris in many of the intestinal crypts (arrows).
This is a higher-power photomicrograph showing the eosinophilic debris in many of the intestinal crypts (arrows). -
 This higher-power photomicrograph shows more clearly the eosinophilic debris (arrows) in the intestinal crypts.
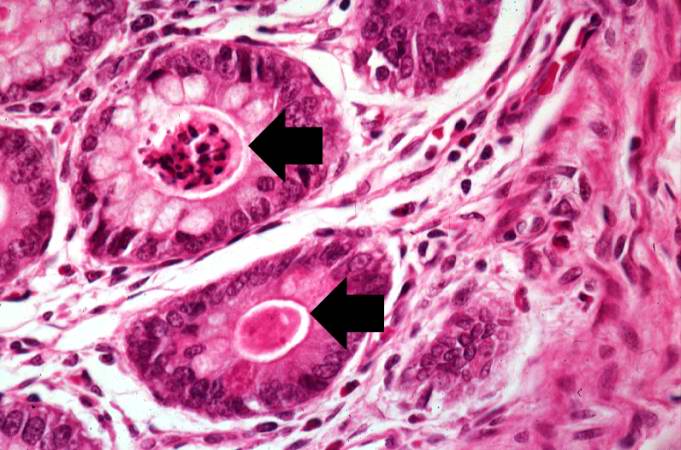
This higher-power photomicrograph shows more clearly the eosinophilic debris (arrows) in the intestinal crypts. -
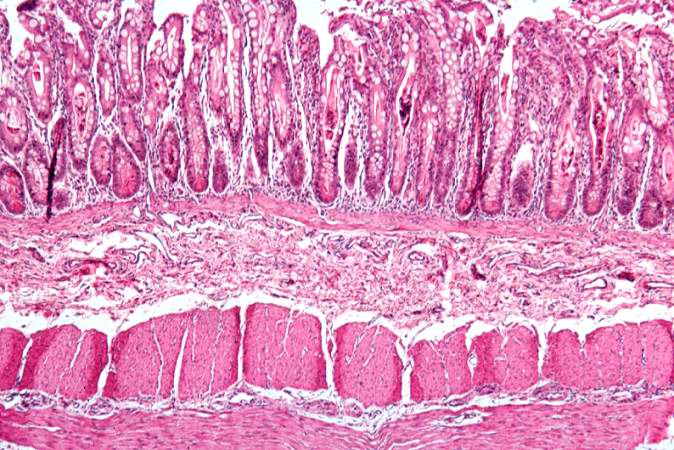 This is a low-power photomicrograph from another section of the intestine. Saggital sections of the intestinal crypts show the crypts along their full length, extending to the mucosal surface.
This is a low-power photomicrograph from another section of the intestine. Saggital sections of the intestinal crypts show the crypts along their full length, extending to the mucosal surface.



-
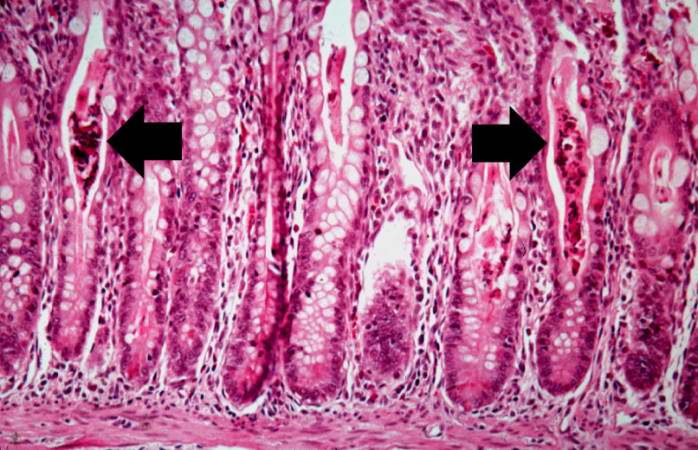 A higher-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows).
A higher-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows). -
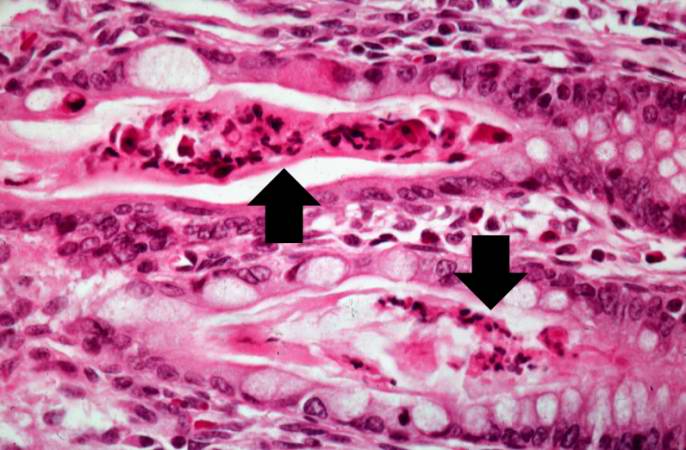 Another high-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows).
Another high-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows). -
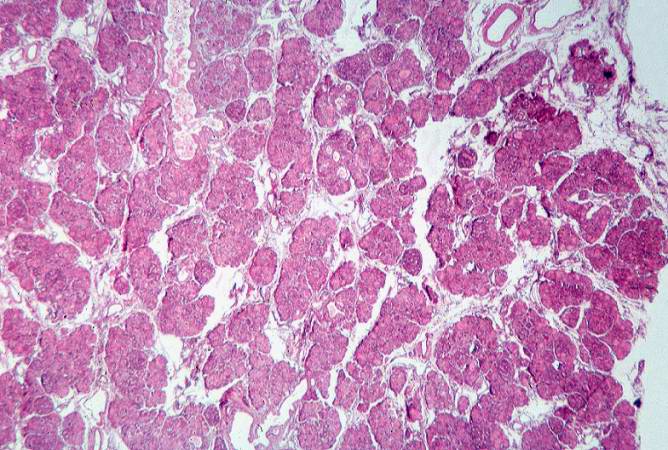 This low-power photomicrograph of pancreas shows increased interstitial connective tissue resulting in accentuation of the lobular pattern.
This low-power photomicrograph of pancreas shows increased interstitial connective tissue resulting in accentuation of the lobular pattern.



References
- ↑ National Center for Biotechnology Information (US). Genes and Disease [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 1998-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22183/
- ↑ Voter KZ, Ren CL (2008). “Diagnosis of cystic fibrosis”. Clin Rev Allergy Immunol. 35 (3): 100–6. doi:10.1007/s12016-008-8078-x. PMID 18506640.
- ↑ 3.0 3.1 3.2 Ratjen FA (2009). “Cystic fibrosis: pathogenesis and future treatment strategies”. Respir Care. 54 (5): 595–605. PMID 19393104.
- ↑ 4.0 4.1 4.2 Cutting GR (2015). “Cystic fibrosis genetics: from molecular understanding to clinical application”. Nat. Rev. Genet. 16 (1): 45–56. doi:10.1038/nrg3849. PMC 4364438. PMID 25404111.
- ↑ Konstan MW, Ratjen F (2012). “Effect of dornase alfa on inflammation and lung function: potential role in the early treatment of cystic fibrosis”. J. Cyst. Fibros. 11 (2): 78–83. doi:10.1016/j.jcf.2011.10.003. PMC 4090757. PMID 22093951.
- ↑ 6.0 6.1 Ratjen F, Döring G (2003). “Cystic fibrosis”. Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185.
- ↑ “Cystic Fibrosis – National Library of Medicine – PubMed Health”.
- ↑ “File:CFTR.jpg – Wikimedia Commons”. External link in
|title=(help) - ↑ Prevalence of ΔF508, G551D, G542X, R553X mutations among cystic fibrosis patients in the North of Brazil. Brazilian Journal of Medical and Biological Research 2005; 38:11–15. PMID 15665983
- ↑ Ratjen F, Döring G (2003). “Cystic fibrosis”. Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185.
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
Cystic fibrosis is caused by mutations in the CFTR gene. The genetic mutations result in defective transport of chloride, and secondarily sodium, by epithelial cells and eventually abnormal viscous mucoid secretions mostly in lungs and GI tract.
Causes
Genetic Cause
- Cystic fibrosis is caused by mutations in the CFTR gene. The genetic mutations result in defective transport of chloride, and secondarily sodium, by epithelial cells and eventually abnormal viscous mucoid secretions mostly in lungs and GI tract.[1]
References
- ↑ Hull J (June 2012). “Cystic fibrosis transmembrane conductance regulator dysfunction and its treatment”. J R Soc Med. 105 Suppl 2: S2–8. doi:10.1258/jrsm.2012.12s001. PMC 3372304. PMID 22688363.
Differentiating Cystic fibrosis from other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2], Karina Zavaleta, MD [3], Anmol Pitliya, M.B.B.S. M.D.[4]
Overview
Cystic fibrosis has to be differentiated from other conditions with similar presentation of cough and wheeze like common cold, asthma, bronchiolitis, emphysema, primary ciliary dyskinesia (Kartagener syndrome), bronchitis, bronchiectasis, foreign body aspiration, pneumoconiosis, interstitial lung disease, cardiogenic pulmonary edema, GERD and sarcoidosis.
Cough
Cystic fibrosis must be differentiated from other diseases presenting with cough and wheeze include:
| Organ system | Diseases | Clinical manifestations | Diagnosis | Other features | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Symptoms | Physical exam | ||||||||||||||
| Onset | Duration | Productive cough | Hemoptysis | Weight lost | Fever | Dyspnea | Ascultation | Lab findings | Imaging | PFT | Gold standard | ||||
| Respiratory | Upper airway diseases | Common Cold[1] | Acute |
|
✔ | – | – | ✔ | – |
|
|
|
|
||
| Lower airway | Asthma[2] | Chronic |
|
✔ Clear mucoid or yellow sputum | – | – | – | ✔ |
|
|
|
|
| ||
| Acute Bronchitis[3] | Acute |
|
✔ | – | – | – | ✔ |
|
|
|
|
| |||
| Chronic Bronchitis[4][5] | Chronic |
|
✔ Clear sputum | – | – | ✔ | ✔ |
|
|
|
| ||||
| Primary Ciliary Dyskinesia | Chronic |
|
✔ | ✔ | ✔ | – | ✔ |
|
|
|
|
| |||
| Bronchiectasis[6] | Chronic |
|
✔ Mucopurulent sputum | ✔ | – | ✔ |
|
|
|
| |||||
| Emphysema [7] | Chronic |
|
✔ Mucoid or purulent sputum | – | – | ✔ | ✔ |
|
|
|
|
| |||
| Foreign body aspiration[8][9][10] | Acute |
|
✔ | ✔ | – | ✔ | ✔ |
|
|
|
|
| |||
| Bronchiolitis[11][12] | Acute |
|
✔ | – | ✔ | ✔ |
|
|
|
|
| ||||
| Parenchyma | Cystic fibrosis [13][14] | Chronic |
|
✔ | – | ✔ | ✔ |
|
|
| |||||
| Pneumoconioses[15][16] | Acute, Chronic |
|
– | – | ✔ | ✔ | ✔ |
|
|
||||||
| Interstitial lung disease[17][18] | Chronic |
|
– | ✔ | ✔ | – | ✔ |
|
|
|
|
| |||
| Cardiac | Cardiogenic pulmonary edema[19][20] | Acute |
|
✔ Pink frothy, liquid | – | ✔ | – | ✔ |
|
|
|
|
| ||
| Gastrointestinal | Gastroesophageal reflux[21][22] | Chronic |
|
✔ | – | ✔ | – | ✔ |
|
|
|
|
— | ||
| Autoinmune | Sarcoidosis[23][24] | Chronic |
|
– | – | ✔ | ✔ | ✔ |
|
|
|
||||
References
- ↑ Eccles R (2005). “Understanding the symptoms of the common cold and influenza”. Lancet Infect Dis. 5 (11): 718–25. doi:10.1016/S1473-3099(05)70270-X. PMID 16253889.
- ↑ Ukena D, Fishman L, Niebling WB (2008). “Bronchial asthma: diagnosis and long-term treatment in adults”. Dtsch Arztebl Int. 105 (21): 385–94. doi:10.3238/arztebl.2008.0385. PMC 2696883. PMID 19626179.
- ↑ Wenzel RP, Fowler AA (2006). “Clinical practice. Acute bronchitis”. N. Engl. J. Med. 355 (20): 2125–30. doi:10.1056/NEJMcp061493. PMID 17108344.
- ↑ Brusasco V, Martinez F (2014). “Chronic obstructive pulmonary disease”. Compr Physiol. 4 (1): 1–31. doi:10.1002/cphy.c110037. PMID 24692133.
- ↑ Qaseem A, Snow V, Shekelle P, Sherif K, Wilt TJ, Weinberger S, Owens DK (2007). “Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline from the American College of Physicians”. Ann. Intern. Med. 147 (9): 633–8. PMID 17975186.
- ↑ King PT, Holdsworth SR, Freezer NJ, Villanueva E, Holmes PW (2006). “Characterisation of the onset and presenting clinical features of adult bronchiectasis”. Respir Med. 100 (12): 2183–9. doi:10.1016/j.rmed.2006.03.012. PMID 16650970.
- ↑ Rossi A, Butorac-Petanjek B, Chilosi M, Cosío BG, Flezar M, Koulouris N; et al. (2017). “Chronic obstructive pulmonary disease with mild airflow limitation: current knowledge and proposal for future research – a consensus document from six scientific societies”. Int J Chron Obstruct Pulmon Dis. 12: 2593–2610. doi:10.2147/COPD.S132236. PMC 5587130. PMID 28919728.
- ↑ Hewlett JC, Rickman OB, Lentz RJ, Prakash UB, Maldonado F (2017). “Foreign body aspiration in adult airways: therapeutic approach”. J Thorac Dis. 9 (9): 3398–3409. doi:10.21037/jtd.2017.06.137. PMC 5708401. PMID 29221325.
- ↑ Rafanan AL, Mehta AC (2001). “Adult airway foreign body removal. What’s new?”. Clin. Chest Med. 22 (2): 319–30. PMID 11444115.
- ↑ Haddadi S, Marzban S, Nemati S, Ranjbar Kiakelayeh S, Parvizi A, Heidarzadeh A (2015). “Tracheobronchial Foreign-Bodies in Children; A 7 Year Retrospective Study”. Iran J Otorhinolaryngol. 27 (82): 377–85. PMC 4639691. PMID 26568942.
- ↑ Bordley WC, Viswanathan M, King VJ, Sutton SF, Jackman AM, Sterling L, Lohr KN (2004). “Diagnosis and testing in bronchiolitis: a systematic review”. Arch Pediatr Adolesc Med. 158 (2): 119–26. doi:10.1001/archpedi.158.2.119. PMID 14757603.
- ↑ “www.nice.org.uk”.
- ↑ Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW (2008). “Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report”. J. Pediatr. 153 (2): S4–S14. doi:10.1016/j.jpeds.2008.05.005. PMC 2810958. PMID 18639722.
- ↑ Kerem E, Reisman J, Corey M, Canny GJ, Levison H (1992). “Prediction of mortality in patients with cystic fibrosis”. N. Engl. J. Med. 326 (18): 1187–91. doi:10.1056/NEJM199204303261804. PMID 1285737.
- ↑ Jp NA, Imanaka M, Suganuma N (2017). “Japanese workplace health management in pneumoconiosis prevention”. J Occup Health. 59 (2): 91–103. doi:10.1539/joh.16-0031-RA. PMC 5478517. PMID 27980247.
- ↑ Weiland DA, Lynch DA, Jensen SP, Newell JD, Miller DE, Crausman RS, Kuhn C, Kern DG (2003). “Thin-section CT findings in flock worker’s lung, a work-related interstitial lung disease”. Radiology. 227 (1): 222–31. doi:10.1148/radiol.2271011063. PMID 12668748.
- ↑ Lama VN, Martinez FJ (2004). “Resting and exercise physiology in interstitial lung diseases”. Clin. Chest Med. 25 (3): 435–53, v. doi:10.1016/j.ccm.2004.05.005. PMID 15331185.
- ↑ Chetta A, Marangio E, Olivieri D (2004). “Pulmonary function testing in interstitial lung diseases”. Respiration. 71 (3): 209–13. doi:10.1159/000077416. PMID 15133338.
- ↑ Gheorghiade M, Zannad F, Sopko G, Klein L, Piña IL, Konstam MA, Massie BM, Roland E, Targum S, Collins SP, Filippatos G, Tavazzi L (2005). “Acute heart failure syndromes: current state and framework for future research”. Circulation. 112 (25): 3958–68. doi:10.1161/CIRCULATIONAHA.105.590091. PMID 16365214.
- ↑ Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL (2013). “2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines”. Circulation. 128 (16): e240–327. doi:10.1161/CIR.0b013e31829e8776. PMID 23741058.
- ↑ Kahrilas PJ, Hughes N, Howden CW (2011). “Response of unexplained chest pain to proton pump inhibitor treatment in patients with and without objective evidence of gastro-oesophageal reflux disease”. Gut. 60 (11): 1473–8. doi:10.1136/gut.2011.241307. PMID 21508423.
- ↑ Badillo R, Francis D (2014). “Diagnosis and treatment of gastroesophageal reflux disease”. World J Gastrointest Pharmacol Ther. 5 (3): 105–12. doi:10.4292/wjgpt.v5.i3.105. PMC 4133436. PMID 25133039.
- ↑ Carmona EM, Kalra S, Ryu JH (2016). “Pulmonary Sarcoidosis: Diagnosis and Treatment”. Mayo Clin. Proc. 91 (7): 946–54. doi:10.1016/j.mayocp.2016.03.004. PMID 27378039.
- ↑ Yanardağ H, Pamuk GE, Karayel T, Demirci S (2002). “Bone marrow involvement in sarcoidosis: an analysis of 50 bone marrow samples”. Haematologia (Budap). 32 (4): 419–25. PMID 12803116.
Epidemiology and Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
The incidence of cystic fibrosis is approximately 1 in 2500 livebirths. It is a life-limiting disease (100% mortality rate), and a cure for the disease remains elusive. Most patients with cystic fibrosis are diagnosed in first 2 years of life. The onset of symptoms is before the first month of life in 12%, between 1-6 months of age in 75%, and between 6-12 months of age in 7% of patients. Although cystic fibrosis has been reported in all racial and ethnic groups, it mostly affects Caucasians of Northern European descent. It affects men and women equally.
Epidemiology and Demographics
Incidence
- The incidence of cystic fibrosis is approximately 40 in 100,000 live births worldwide.
- The incidence of cystic fibrosis is approximately 25 in 100,000 newborns in the US.[1][2]
Prevalence
- Cystic fibrosis affects more than 30,000 people in the United States and 80,000 people worldwide.[3]
Mortality rate
- Cystic fibrosis is a life-limiting disease (100% mortality rate), and a cure for the disease remains elusive.[4]
Age
- Most patients with cystic fibrosis are diagnosed in first 2 years of life. The onset of symptoms is before the first month of life in 12%, between 1-6 months of age in 75%, and between 6-12 months of age in 7% of patients.[5]
Race
- Although cystic fibrosis has been reported in all racial and ethnic groups, it mostly affects Caucasians of Northern European descent.
- Cystic fibrosis is the most lethal genetic disease among Caucasians
- Cystic fibrosis is approximately reported in different ethnic groups as follow:[6][7]
| Prevalence of cystic fibrosis according to race | |
|---|---|
| Race | Prevalence (per 100,000 person years) |
| Caucasian | 40 |
| Hispanic | 7.4 |
| African-American | 6.6 |
Gender
- Cystic fibrosis affects men and women equally. Women with cystic fibrosis have a shortened life expectancy compared to men.[8]
References
- ↑ Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, Howenstine M, McColley SA, Rock M, Rosenfeld M, Sermet-Gaudelus I, Southern KW, Marshall BC, Sosnay PR (February 2017). “Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation”. J. Pediatr. 181S: S4–S15.e1. doi:10.1016/j.jpeds.2016.09.064. PMID 28129811.
- ↑ Ratjen F, Döring G (2003). “Cystic fibrosis”. Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185.
- ↑ Brown SD, White R, Tobin P (May 2017). “Keep them breathing: Cystic fibrosis pathophysiology, diagnosis, and treatment”. JAAPA. 30 (5): 23–27. doi:10.1097/01.JAA.0000515540.36581.92. PMID 28441669.
- ↑ Pittman JE, Ferkol TW (August 2015). “The Evolution of Cystic Fibrosis Care”. Chest. 148 (2): 533–542. doi:10.1378/chest.14-1997. PMC 4524331. PMID 25764168.
- ↑ Ernst MM, Johnson MC, Stark LJ (April 2010). “Developmental and psychosocial issues in cystic fibrosis”. Child Adolesc Psychiatr Clin N Am. 19 (2): 263–83, viii. doi:10.1016/j.chc.2010.01.004. PMC 2874200. PMID 20478499.
- ↑ Pettit RS, Fellner C (July 2014). “CFTR Modulators for the Treatment of Cystic Fibrosis”. P T. 39 (7): 500–11. PMC 4103577. PMID 25083129.
- ↑ Cutting GR (2015). “Cystic fibrosis genetics: from molecular understanding to clinical application”. Nat. Rev. Genet. 16 (1): 45–56. doi:10.1038/nrg3849. PMC 4364438. PMID 25404111.
- ↑ Harness-Brumley CL, Elliott AC, Rosenbluth DB, Raghavan D, Jain R (December 2014). “Gender differences in outcomes of patients with cystic fibrosis”. J Womens Health (Larchmt). 23 (12): 1012–20. doi:10.1089/jwh.2014.4985. PMC 4442553. PMID 25495366.
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
Every individual with cystic fibrosis inherits two CFTR genes, one from each parent. Children who inherit two mutated CFTR genes from both parents will have cystic fibrosis.
Risk Factors
- Every individual with cystic fibrosis inherits two CFTR genes, one from each parent. Children who inherit two mutated CFTR genes from both parents will have cystic fibrosis.[1]
- Children with only one gene will act as a carrier for next generations.
References
Screening
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
Newborn screening identified most of the children with cystic fibrosis before the symptoms develop. It offers this opportunity for early diagnosis and improved outcomes. Immunoreactive trypsinogen (IRT) of serum is raised in newborns with cystic fibrosis and has been used as a screening test. A raised IRT in the first week of life is a sensitive test but not specific for cystic fibrosis.
Screening
- In almost all of the Western countries, newborn screening for cystic fibrosis (CF NBS) has been introduced that identified most of the children with cystic fibrosis before the symptoms develop.
- It offers this opportunity for early diagnosis and improved outcomes in patients with cystic fibrosis.[1][2]
- Immunoreactive trypsinogen (IRT) of serum is raised in newborns with cystic fibrosis and has been used as a screening test.
- A raised IRT in the first week of life (b-IRT) is a sensitive test but not specific for cystic fibrosis. A second IRT test within 6 weeks of life is used in some screening protocols to avoid excessive numbers of sweat chloride test.[3][4]
References
- ↑ Gonska T, Ratjen F (October 2015). “Newborn screening for cystic fibrosis”. Expert Rev Respir Med. 9 (5): 619–31. doi:10.1586/17476348.2015.1085804. PMID 26366807.
- ↑ Hale JE, Parad RB, Dorkin HL, Gerstle R, Lapey A, O’Sullivan BP, Spencer T, Yee W, Comeau AM (October 2010). “Cystic fibrosis newborn screening: using experience to optimize the screening algorithm”. J. Inherit. Metab. Dis. 33 (Suppl 2): S255–61. doi:10.1007/s10545-010-9117-3. PMID 20521170.
- ↑ Rock MJ, Mischler EH, Farrell PM, Bruns WT, Hassemer DJ, Laessig RH (1989). “Immunoreactive trypsinogen screening for cystic fibrosis: characterization of infants with a false-positive screening test”. Pediatr. Pulmonol. 6 (1): 42–8. PMID 2704582.
- ↑ Paracchini V, Seia M, Raimondi S, Costantino L, Capasso P, Porcaro L, Colombo C, Coviello DA, Mariani T, Manzoni E, Sangiovanni M, Corbetta C (2012). “Cystic fibrosis newborn screening: distribution of blood immunoreactive trypsinogen concentrations in hypertrypsinemic neonates”. JIMD Rep. 4: 17–23. doi:10.1007/8904_2011_55. PMC 3509858. PMID 23430892.
Natural History, Complications and Prognosis
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] ; Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
Malnutrition and poor growth (due to loss of pancreatic exocrine function) leads to death in the first decade of life for most untreated patients. The most significant complications are seen in airways (responsible for 80% of mortality) and most common chronic pulmonary infection include P. aeruginosa, S. aureus and H. influenzae. In cystic fibrosis 98% of men are infertile due to aspermia. Lung complications are currently the primary causes of morbidity and are responsible for 80% of mortality in these patients and gastrointestinal complications include pancreatic insufficiency, pancreatitis, gastroesophageal reflux disease, distal intestinal obstuction syndrome, constipation and small intestinal bacterial overgrowth. In cystic fibrosis, obstructive lung disease and other lung complications are currently the primary causes of morbidity and are responsible for 80% of mortality. At present time survival probability of children is 40-50 years. Women with cystic fibrosis have a shortened life expectancy compared to men.
Natural History
- If patients with cystic fibrosis left untreated, they would present with failure to thrive and/or steatorrhea during infancy. Also this would typically progress to hypoproteinemia, edema, and severe cachexia.[1]
- Malnutrition and poor growth (due to loss of pancreatic exocrine function) leads to death in the first decade of life for most untreated patients.[2]
Complications
Lung involvement:
In patients with cystic fibrosis the most significant changes and complications are seen in the airways. The primary genetic defect eventually causes chronic pulmonary infections. P. aeruginosa is the most common infection, followed by S. aureus and H. influenzae.[3][4]
- Infancy: the most common bacteria cultured is S. aureus along with H. influenzae recently increased during childhood
- Adolescence and young adulthood: the commonest pathogen cultured is P. aeruginosa
Other lung complications of cystic fibrosis include:[5]
Gastrointestinal involvement:
In cystic fibrosis, approximately 90% of patients present with exocrine pancreatic insufficiency. Pancreatic insufficiency leads to maldigestion and malabsorption of nutrients, followed by sequelae of malnutrition include permanent stunting of stature, cognitive dysfunction (due to vitamin E deficiency) and more rapid decline in pulmonary function. Other gastrointestinal complications related to cystic fibrosis include:[5][6][7]
- Pancreatitis
- Gastroesophageal reflux disease
- Distal intestinal obstuction syndrome
- Obstipation/constipation
- Small intestinal bacterial overgrowth
- Steatosis
- Cholelithiasis
- Meconium ileus
- Malabsorption (vitamin malabsorption may cause hemolytic anemia and defective coagulation)
Endocrine system invovement:
Endocrine complications related to cystic fibrosis include:[5]
Others
- Salt-loss syndromes (acute salt depletion, chronic metabolic alkalosis, and/or hyponatremic hypochloremic dehydration)
- Hemolytic anemia and defective coagulation due to vitamin malabsorption [8]
Prognosis
- Life expectancy of patients with cystic fibrosis has been increased over past decades because of better symptomatic treatment strategies.
- In patients with cystic fibrosis, obstructive lung disease and other lung complications are currently the primary causes of morbidity and are responsible for 80% of mortality.[2][9]
- At present, the survival probability of children is 40-50 years.[10]
- Women with cystic fibrosis have a shortened life expectancy compared to men. Women also become colonized with certain common CF pathogens earlier and have a lower life expectancy in the setting of respiratory infections.[11]
References
- ↑ Moskowitz SM, Chmiel JF, Sternen DL, Cheng E, Gibson RL, Marshall SG, Cutting GR (December 2008). “Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders”. Genet. Med. 10 (12): 851–68. doi:10.1097/GIM.0b013e31818e55a2. PMC 2810953. PMID 19092437.
- ↑ 2.0 2.1 Cutting GR (2015). “Cystic fibrosis genetics: from molecular understanding to clinical application”. Nat. Rev. Genet. 16 (1): 45–56. doi:10.1038/nrg3849. PMC 4364438. PMID 25404111.
- ↑ Edmondson C, Davies JC (2016). “Current and future treatment options for cystic fibrosis lung disease: latest evidence and clinical implications”. Ther Adv Chronic Dis. 7 (3): 170–83. doi:10.1177/2040622316641352. PMC 4907071. PMID 27347364.
- ↑ Ratjen FA (2009). “Cystic fibrosis: pathogenesis and future treatment strategies”. Respir Care. 54 (5): 595–605. PMID 19393104.
- ↑ 5.0 5.1 5.2 Flume PA (May 2009). “Pulmonary complications of cystic fibrosis”. Respir Care. 54 (5): 618–27. PMID 19393106.
- ↑ Gelfond D, Borowitz D (April 2013). “Gastrointestinal complications of cystic fibrosis”. Clin. Gastroenterol. Hepatol. 11 (4): 333–42, quiz e30–1. doi:10.1016/j.cgh.2012.11.006. PMID 23142604.
- ↑ Sabharwal S (January 2016). “Gastrointestinal Manifestations of Cystic Fibrosis”. Gastroenterol Hepatol (N Y). 12 (1): 43–7. PMC 4865785. PMID 27330503.
- ↑ Moskowitz SM, Chmiel JF, Sternen DL, Cheng E, Gibson RL, Marshall SG, Cutting GR (December 2008). “Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders”. Genet. Med. 10 (12): 851–68. doi:10.1097/GIM.0b013e31818e55a2. PMC 2810953. PMID 19092437.
- ↑ Pettit RS, Fellner C (July 2014). “CFTR Modulators for the Treatment of Cystic Fibrosis”. P T. 39 (7): 500–11. PMC 4103577. PMID 25083129.
- ↑ Fila L (2018). “[Cystic fibrosis in adults]”. Vnitr Lek (in Czech). 63 (11): 834–842. PMID 29303286.
- ↑ Harness-Brumley CL, Elliott AC, Rosenbluth DB, Raghavan D, Jain R (December 2014). “Gender differences in outcomes of patients with cystic fibrosis”. J Womens Health (Larchmt). 23 (12): 1012–20. doi:10.1089/jwh.2014.4985. PMC 4442553. PMID 25495366.
Diagnosis
Diagnosis
Diagnostic Study of Choice | History and Symptoms | Physical Examination | Laboratory Findings | Electrocardiogram | Chest X Ray | Echocardiography or Ultrasound | CT | MRI | Other Imaging Findings | Other Diagnostic Studies
Treatment
Treatment
Medical Therapy | Surgery | Primary Prevention | Secondary Prevention | Cost-Effectiveness of Therapy | Future or Investigational Therapies
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH