
Dilated cardiomyopathy
For patient information, click here
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Abdelrahman Ibrahim Abushouk, MD[2]; Sachin Shah, M.D.
Synonyms and keywords: Congestive cardiomyopathy; DCM
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor-in-Chief: Abdelrahman Ibrahim Abushouk, MD[2]; Sachin Shah, M.D.
Overview
Dilated cardiomyopathy is a condition of the heart that causes dilation and impaired contraction of the left ventricle (or both ventricles). Impaired contraction is defined as a low ejection fraction (< 40%).
Historical Perspective
The etiology of dilated cardiomyopathy remained elusive for a long time that it was defined by the World Health Organization as a “heart muscle disorder of unknown cause”. However, recent research highlighted several genetic mutations that are associated with the condition. Therefore, the more recent definition by the American Heart Association was “a myocardial disorder with mechanical dysfunction, which usually exhibits inappropriate ventricular dilatation, due to a variety of etiologies that frequently are genetic”.
Pathophysiology
Familial traits and mitochondrial inheritance are thought to play a part in the development of idiopathic dilated cardiomyopathy, and the inheritance occurs in an autosomal dominant pattern. Connective tissue disease, and other diseases or toxins that disrupt the tissue of the heart are also implicated in the development of dilated cardiomyopathy.
Causes
There are many causes of dilated cardiomyopathy. The most common cause is idiopathic in 50% of cases. The next most common cause is myocarditis which is responsible for 10% of cases. Other common causes include substance abuse, connective tissue disease, pregnancy, medications, nutritional deficiencies, infiltrative diseases and toxins.
Epidemiology and Demographics
Dilated cardiomyopathy is most likely to occur between the ages of 20-60, is three times as likely to occur in males over females, and is 2.5 times more likely to occur in African Americans.
Screening
The current guidelines recommend screening for dilated cardiomyopathy in individuals with 2 or 3 family members with primary dilated cardiomyopathy. Screening can be performed using electrocardiograms and echocardiography to measure the size and function of the left ventricle. An underlying genetic mutation in the 40 genes (currently assessed in familial dilated cardiomyopathy genetic testing) can be detected in 30 to 40% of DCM patients.
Natural History, Complications and Prognosis
There are several prognostic indicators when evaluating dilated cardiomyopathy, the most important one being ejection fraction. Complications as a result of dilated cardiomyopathy include heart failure, aortic and mitral valve regurgitation, emboli, edema, arrhythmias and sudden cardiac arrest.
Diagnosis
History and Symptoms
Common symptoms in the setting of dilated cardiomyopathy include chest pain, cough, fatigue, loss of appetite, and shortness of breath. A careful history is important in the setting of dilated cardiomyopathy in order to ascertain the etiology of the cardiomyopathy. The patient needs to be evaluated for a history of coronary artery disease, viral prodrome and infections, chemotherapy, HIV risk factors, pregnancy, medications, toxins, and substance abuse.
Laboratory Findings
The majority of dilated cardiomyopathy lab workup is targeted towards detecting the cause (such as thyroid function tests, toxicology screening, and genetic counselling) or assessing the cardiac complications of the condition. Other biomarkers that are under investigation include serum uric acid, Ca-125, soluble ST2, and Growth and differentiation factor-15.
MRI
In patients presenting with heart failure, where the etiology of the cardiac dysfunction is unclear, cardiac MRI can be a useful imaging modality. It can be used to distinguish the area of inflammation, to help in the diagnosis of myocarditis, to evaluate patients with suspected infiltrative diseases, and to evaluate dilated cardiomyopathy in the setting of normal coronary arteries.
Echocardiography
Echocardiography is the most common imaging finding used to diagnose dilated cardiomyopathy. Findings may include ventricular and atrial dilatation, increased left ventricular mass, a global reduction in systolic function, and focal wall motion abnormalities.
Other Diagnostic Studies
Endomyocardial biopsy has low sensitivy and the findings are also notoriously non-specific. The findings on biopsy usually involve findings of inflammation and specific pathogens are unlikely to be identified. There may be an increased yield to using MRI to target endomyocardial biopsy. Viral titiers (serologies) are often unhelpful and not routinely ordered in clinical practice.
Treatment
Medical Therapy
Treatment should focus on correcting the underlying cause of the cardiomyopathy when possible. Treatment is also targeted towards preventing death, and ameliorating the symptoms of heart failure. Medications that have been proven to reduce mortality in patients with systolic heart failure are; ACE inhibitors, beta blockers, angiotensin II receptor blockers, nitrates, and hydralazine. Diuretics and digoxin are used for symptom relief.
Surgery
There are several surgical options for patients with dilated cardiomyopathy, depending on the severity of heart failure. Implantable cardiac defibrillators have been studied in these patient, and may help in preventing arrhythmias. Cardiac transplantation may be an option for patients with severe heart failure, and a left ventricular assist device, or LVAD, may help to bridge a patient while awaiting transplantation. This device may also be used as a palliative measure, called a “destination LVAD” for patients with end-stage heart failure who are not suitable transplant candidates.
References
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]Associate Editor(s)-in-Chief: Abdelrahman Ibrahim Abushouk, MD[2]
Overview
The etiology of dilated cardiomyopathy remained elusive for a long time that it was defined by the World Health Organization as a “heart muscle disorder of unknown cause”. However, recent research highlighted several genetic mutations that are associated with the condition. Therefore, the more recent definition by the American Heart Association was “a myocardial disorder with mechanical dysfunction, which usually exhibits inappropriate ventricular dilatation, due to a variety of etiologies that frequently are genetic”.
Historical Perspective
- In the 1980s, the World Health Organization defined cardiomyopathies as “heart muscle diseases of unknown cause”.[1] This definition reflected the poor understanding of disease etiology.
- Over the past two decades, significant advances have been made in understanding the genetic etiology of dilated cardiomyopathy.
- Mutations in over 80 genes have been associated with this condition.[2]
- Therefore, more recently, the American Heart Association defined cardiomyopathies as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction, which usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation, due to a variety of etiologies that frequently are genetic“.[3]
- Recent studies have revealed the complexity of the genetic basis for dilated cardiomyopathy; however, much remains to be investigated to fully understand the etiology of this condition.
References
- ↑ “Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies”. Br Heart J. 44 (6): 672–3. 1980. doi:10.1136/hrt.44.6.672. PMC 482464. PMID 7459150.
- ↑ Hershberger RE, Hedges DJ, Morales A (2013). “Dilated cardiomyopathy: the complexity of a diverse genetic architecture”. Nat Rev Cardiol. 10 (9): 531–47. doi:10.1038/nrcardio.2013.105. PMID 23900355.
- ↑ Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D; et al. (2006). “Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention”. Circulation. 113 (14): 1807–16. doi:10.1161/CIRCULATIONAHA.106.174287. PMID 16567565.
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Sachin Shah, M.D.; Jennifer Hall; Abdelrahman Ibrahim Abushouk, MD[2]
Overview
Cardiomyopathies are defined as a heterogeneous group of diseases of the heart associated with a mechanical and/or electrical dysfunction that usually (but not always) exhibit inappropropriate ventricular hypertrophy or dilation and are due to a variety of causes that frequently are genetic. Phenotypic characteristics typically include ventricular chamber enlargement and systolic dysfunction with normal wall thickness. Patients with dilated cardiomyopathy may experience a progressive decline in left ventricular contractile function, ventricular and supraventricular arrhythmias, conduction system problems, thromboembolism, sudden cardiac death and/or heart failure. Dilated cardiomyopathy is the third most common cause of heart failure.
Pathophysiology
Physiology
The normal physiology of myocardium can be understood as follows:
- The myocardium is composed of specialized cardiac muscle cells with an ability not possessed by muscle tissue elsewhere in the body. Cardiac muscle, like other muscles, can contract, but it can also carry an action potential (i.e. conduct electricity), like the neurones that constitute nerves.
- The cardiac myocyte is a specialized muscle cell, which is composed of bundles of myofibrils that contain myofilaments. The myofibrils have distinct micro-anatomical units, called “sarcomeres“, which are considered as the basic contractile units of the cardiac cell. The sarcomere is defined as the region of myofilament structures between two Z-lines. The distance between Z-lines ranges between 1.6 and 2.2 μ. The sarcomere is composed of thick (myosin) and thin (actin) filaments. The chemical and physical interactions between the actin and myosin shortens the sarcomere length and the myocyte to contract during the process of excitation-contraction coupling, which is known as the “sliding filament theory of muscle contraction“.[1]

Pathogenesis
Dilated cardiomyopathy usually results from a failed physiological response to myocyte injury. Mocyte injury can generally end in one of three outcomes: Immediate myocyte cell death, delayed myocyte cell death (apoptosis), or pathological compensatory response.[2] The third outcome usually results in a cycle that occurs as follows:
- Myocyte injury
- Hypertrophy of the remaining myocytes to increased wall stress
- Hyperadrenergic response
- Dynamic remodeling of the interstitial myocardial skeleton (e.g. fibrosis).
- Reduced diastolic function and increased ventricular dilatation.
- Distortion of valvular apparatus
- Increased ventricular afterload
- Initiating the process of heart failure that causes more myocyte injury.[3]
Genetics
Our understanding of the role of genetics in dilated cardiomyopathy continues to grow. Inherited familial dilated cardiomyopathy has been associated with 50 mutations in genes encoding cytoskeletal, nucleoskeletal, mitochondrial and calcium handling proteins.[4] These mutations are listed below.
Genes Encoding Plasma Membrane Proteins
| Gene | Abbreviation |
| Laminin alpha 4 | LAMA4[5] |
| Sarcoglycan delta | SGCD[6][7] |
Genes Encoding Cytoskeletal Proteins
Genes Encoding Calcium Handling Proteins
| Gene | Abbreviation |
| Phospholamban | PLN[66][67][68][69][70][71][72][73] |
Genes Encoding Mitochondrial Proteins
| Gene | Abbreviation |
| Succinate dehydrogenase complex, subunit A, flavoprotein | SDHA[74] |
Genes Encoding Nuclear Proteins
| Gene | Abbreviation |
| ATP-binding cassette, sub-family C, member 9 | ABCC9[75] |
| Lamin A/C | LMNA[76][77][78][79][80][81][82][83] |
| Spectrin repeat containing, nuclear envelope 2 | SYNE2[84] |
The increase in whole exome and whole genome sequencing has significantly increased the number of rare variants that are associated with dilated cardiomyopathy [4]. A challenge in the field today is that many individuals without disease carry rare variants in their genome. Thus the task at hand is not in the sequencing but rather in the translation to define if the rare variants discovered are in fact pathophysiologic in nature. Secondly, evidence is accumulating that many patients with dilated cardiomyopathy may have many different mutations that contribute to or modify disease. [85]
Associated Conditions
A review of systems is also helpful in regards to connective tissue disease associated dilated cardiomyopathy. Some of the disease that can be associated with dilated cardiomyopathy are:
- Systemic lupus erythematosis
- Rheumatoid arthritis
- Sarcoidosis
- Scleroderma
- Connective tissue disease
- Pericardial effusion – It may accompany myocarditis but this finding is not specific.
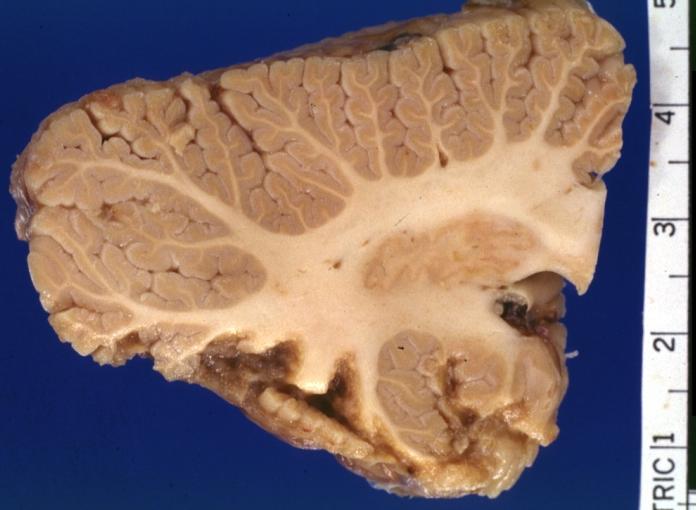
Gross Pathology
On gross pathological examination, the heart may show
- Globular heart (markedly dilated ventricles > 4 cm at the level of papillary muscles)
- Patchy fibrosis in the epicardium
- Endocardial thickening (Cardiac fibroelastosis)
- Ballooning of valve leaflets into the atria
- Few patients show left ventricular non-compaction or minimally dilated ventricles.
Images shown below are Courtesy of Professor Peter Anderson DVM PhD and published with permission. © PEIR, University of Alabama at Birmingham, Department of Pathology
-
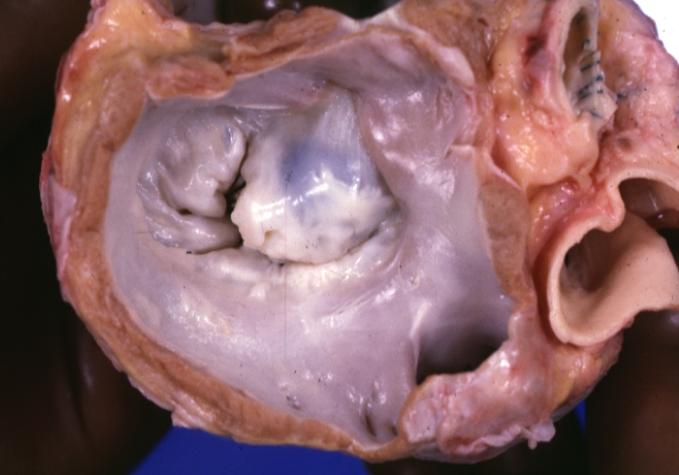 Cardiomyopathy: Gross view from the left atrium, in which the mitral valve anterior leaflet appears to balloon a bit into the atrium
Cardiomyopathy: Gross view from the left atrium, in which the mitral valve anterior leaflet appears to balloon a bit into the atrium -
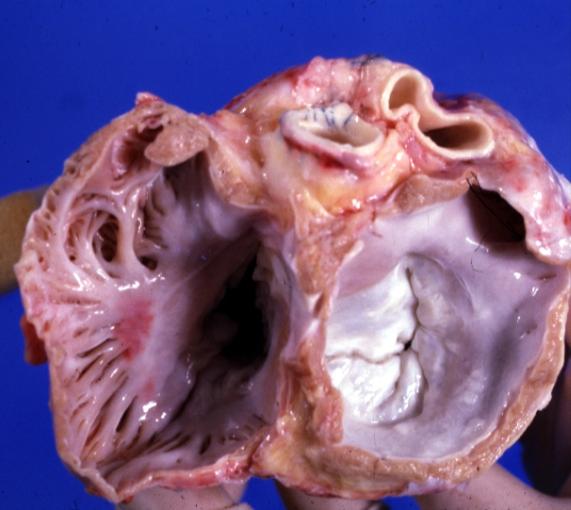 Cardiomyopathy: Gross view of mitral and tricuspid valves from the atria, showing normal anatomy.
Cardiomyopathy: Gross view of mitral and tricuspid valves from the atria, showing normal anatomy.


-
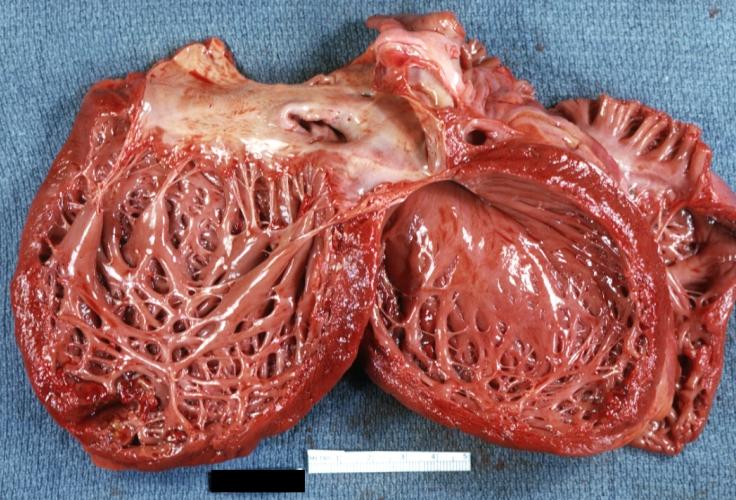
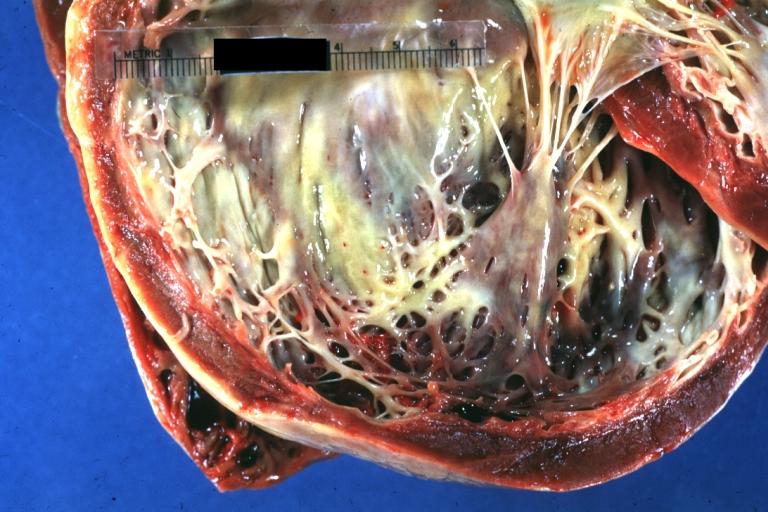 Cardiomyopathy: Gross dilated left ventricle with marked endocardial thickening “adult fibroelastosis”
Cardiomyopathy: Gross dilated left ventricle with marked endocardial thickening “adult fibroelastosis” -
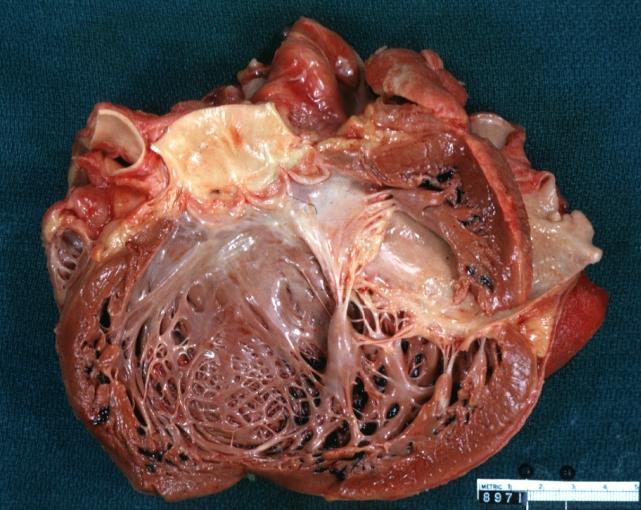 Dilated Cardiomyopathy: Gross dilated left ventricle
Dilated Cardiomyopathy: Gross dilated left ventricle


-
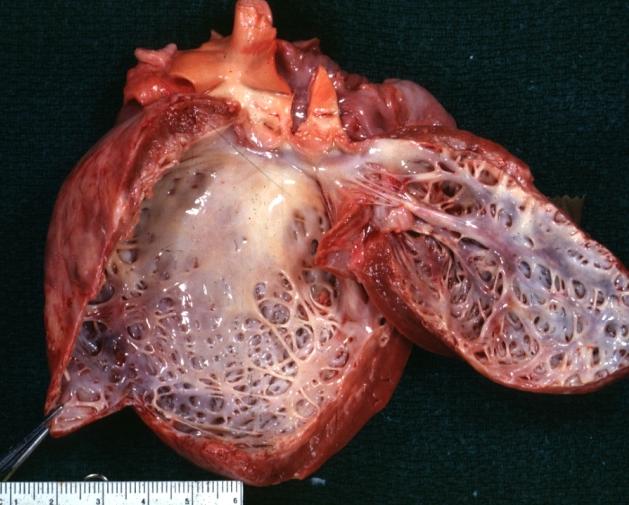 Dilated Cardiomyopathy: Gross dilated left ventricle with marked endocardial sclerosis
Dilated Cardiomyopathy: Gross dilated left ventricle with marked endocardial sclerosis -
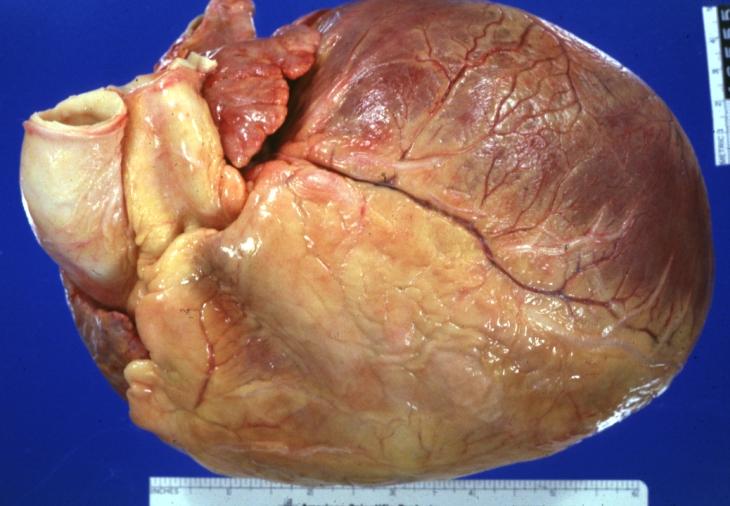 Cardiomyopathy: Gross intact globular shaped heart
Cardiomyopathy: Gross intact globular shaped heart


-
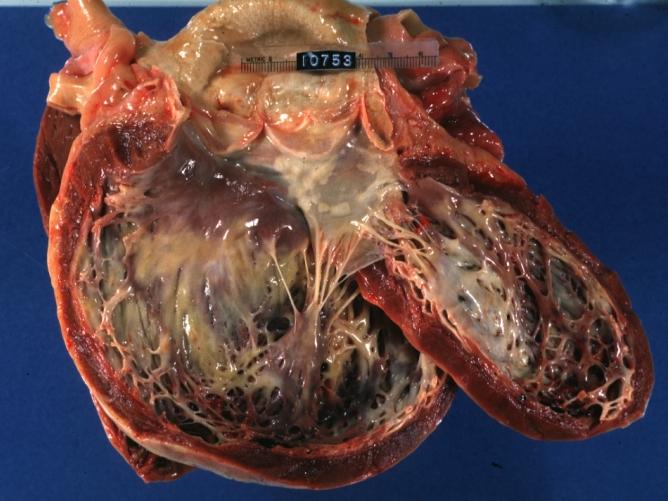 Dilated Cardiomyopathy: Gross opened dilated left ventricle with endocardial thickening
Dilated Cardiomyopathy: Gross opened dilated left ventricle with endocardial thickening -
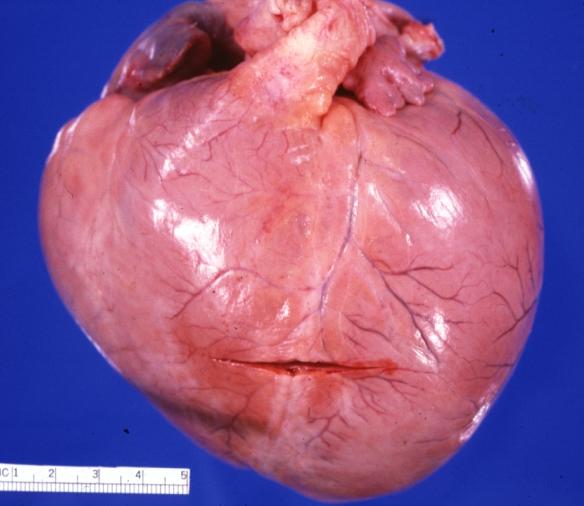 Cardiomyopathy: Gross globular heart (external view) in a 10-year old girl with sickle cell anemia
Cardiomyopathy: Gross globular heart (external view) in a 10-year old girl with sickle cell anemia


-
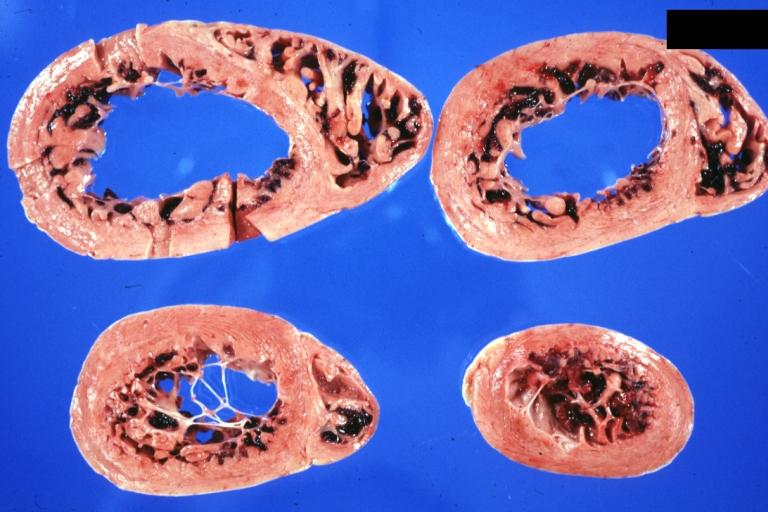 Cardiomyopathy: Gross horizontal sections of ventricles dilation type 10 year old girl with sickle cell anemia
Cardiomyopathy: Gross horizontal sections of ventricles dilation type 10 year old girl with sickle cell anemia -
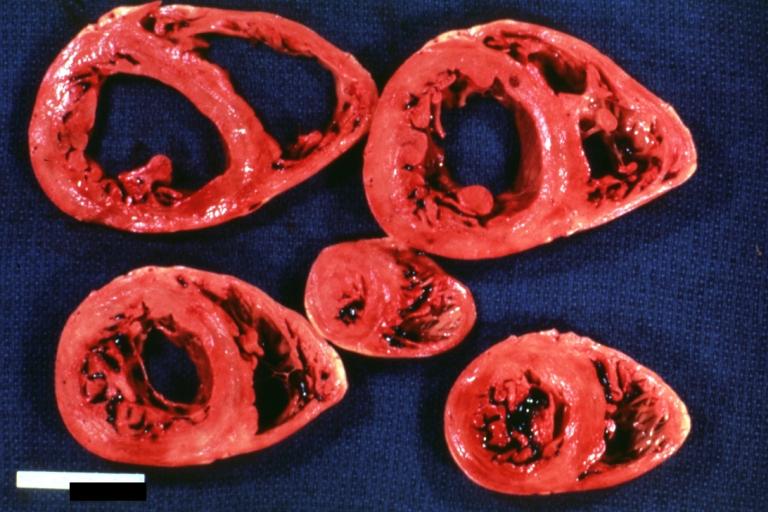 Cardiomyopathy: Intermediate between hypertrophic and dilated
Cardiomyopathy: Intermediate between hypertrophic and dilated


-
 Dilated Cardiomyopathy: Gross opened globular left ventricle
Dilated Cardiomyopathy: Gross opened globular left ventricle

Microscopic Pathology
On microscopic pathological examination, the heart may show
- Variations in myocyte size
- Interstitial fibrosis
- Myofiber disarray
- Transmural scars may be present.
- Further, microscopic examination can verify the underlying cause as inflammation, amyloid, iron, and granulomas.[86]
References
- ↑ Janssen PM (2010). “Myocardial contraction-relaxation coupling”. Am J Physiol Heart Circ Physiol. 299 (6): H1741–9. doi:10.1152/ajpheart.00759.2010. PMC 3006276. PMID 20852049.
- ↑ Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE; et al. (2019). “Dilated cardiomyopathy”. Nat Rev Dis Primers. 5 (1): 32. doi:10.1038/s41572-019-0084-1. PMID 31073128.
- ↑ Weintraub RG, Semsarian C, Macdonald P (2017). “Dilated cardiomyopathy”. Lancet. 390 (10092): 400–414. doi:10.1016/S0140-6736(16)31713-5. PMID 28190577.
- ↑ 4.0 4.1 McNally EM, Golbus JR, Puckelwartz MJ (2013). “Genetic mutations and mechanisms in dilated cardiomyopathy”. J Clin Invest. 123 (1): 19–26. doi:10.1172/JCI62862. PMC 3533274. PMID 23281406.
- ↑ Knöll R, Postel R, Wang J, Krätzner R, Hennecke G, Vacaru AM; et al. (2007). “Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells”. Circulation. 116 (5): 515–25. doi:10.1161/CIRCULATIONAHA.107.689984. PMID 17646580.
- ↑ 6.0 6.1 {{cite journal| author=Tsubata S, Bowles KR, Vatta M, Zintz C, Titus J, Muhonen L et al.| title=Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. | journal=J Clin Invest | year= 2000 | volume= 106 | issue= 5 | pages= 655-62 | pmid=10974018 | doi=10.1172/JCI9224 | pmc=PMC381284 | url=http://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=10974018
- ↑ 7.0 7.1 Trabelsi M, Kavian N, Daoud F, Commere V, Deburgrave N, Beugnet C; et al. (2008). “Revised spectrum of mutations in sarcoglycanopathies”. Eur J Hum Genet. 16 (7): 793–803. doi:10.1038/ejhg.2008.9. PMID 18285821.
- ↑ Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT (1998). “Actin mutations in dilated cardiomyopathy, a heritable form of heart failure”. Science. 280 (5364): 750–2. PMID 9563954.
- ↑ 9.0 9.1 Mohapatra B, Jimenez S, Lin JH, Bowles KR, Coveler KJ, Marx JG; et al. (2003). “Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis”. Mol Genet Metab. 80 (1–2): 207–15. PMID 14567970.
- ↑ Duboscq-Bidot L, Charron P, Ruppert V, Fauchier L, Richter A, Tavazzi L; et al. (2009). “Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy”. Eur Heart J. 30 (17): 2128–36. doi:10.1093/eurheartj/ehp225. PMID 19525294.
- ↑ Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Züchner S; et al. (2011). “Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy”. Am J Hum Genet. 88 (3): 273–82. doi:10.1016/j.ajhg.2011.01.016. PMC 3059419. PMID 21353195.
- ↑ Erdmann J, Hassfeld S, Kallisch H, Fleck E, Regitz-Zagrose V (2000). “Genetic variants in the promoter (g983G>T) and coding region (A92T) of the human cardiotrophin-1 gene (CTF1) in patients with dilated cardiomyopathy”. Hum Mutat. 16 (5): 448. doi:10.1002/1098-1004(200011)16:5<448::AID-HUMU19>3.0.CO;2-D. PMID 11058912.
- ↑ Li D, Tapscoft T, Gonzalez O, Burch PE, Quiñones MA, Zoghbi WA; et al. (1999). “Desmin mutation responsible for idiopathic dilated cardiomyopathy”. Circulation. 100 (5): 461–4. PMID 10430757.
- ↑ Bergman JE, Veenstra-Knol HE, van Essen AJ, van Ravenswaaij CM, den Dunnen WF, van den Wijngaard A; et al. (2007). “Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene”. Eur J Med Genet. 50 (5): 355–66. doi:10.1016/j.ejmg.2007.06.003. PMID 17720647.
- ↑ Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE; et al. (2000). “Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma”. Hum Mol Genet. 9 (18): 2761–6. PMID 11063735.
- ↑ Uzumcu A, Norgett EE, Dindar A, Uyguner O, Nisli K, Kayserili H; et al. (2006). “Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome”. J Med Genet. 43 (2): e5. doi:10.1136/jmg.2005.032904. PMC 2564645. PMID 16467215.
- ↑ Rasmussen TB, Hansen J, Nissen PH, Palmfeldt J, Dalager S, Jensen UB; et al. (2013). “Protein expression studies of desmoplakin mutations in cardiomyopathy patients reveal different molecular disease mechanisms”. Clin Genet. 84 (1): 20–30. doi:10.1111/cge.12056. PMID 23137101.
- ↑ Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P; et al. (2006). “Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition”. J Med Genet. 43 (5): 385–93. doi:10.1136/jmg.2005.036657. PMC 2564511. PMID 16055927.
- ↑ Ferlini A, Galié N, Merlini L, Sewry C, Branzi A, Muntoni F (1998). “A novel Alu-like element rearranged in the dystrophin gene causes a splicing mutation in a family with X-linked dilated cardiomyopathy”. Am J Hum Genet. 63 (2): 436–46. doi:10.1086/301952. PMC 1377294. PMID 9683584.
- ↑ Ortiz-Lopez R, Li H, Su J, Goytia V, Towbin JA (1997). “Evidence for a dystrophin missense mutation as a cause of X-linked dilated cardiomyopathy”. Circulation. 95 (10): 2434–40. PMID 9170407.
- ↑ Todorova A, Constantinova D, Kremensky I (2003). “Dilated cardiomyopathy and new 16 bp deletion in exon 44 of the Dystrophin gene: the possible role of repeated motifs in mutation generation”. Am J Med Genet A. 120A (1): 5–7. doi:10.1002/ajmg.a.10264. PMID 12794683.
- ↑ Milasin J, Muntoni F, Severini GM, Bartoloni L, Vatta M, Krajinovic M; et al. (1996). “A point mutation in the 5′ splice site of the dystrophin gene first intron responsible for X-linked dilated cardiomyopathy”. Hum Mol Genet. 5 (1): 73–9. PMID 8789442.
- ↑ Muntoni F, Cau M, Ganau A, Congiu R, Arvedi G, Mateddu A; et al. (1993). “Brief report: deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy”. N Engl J Med. 329 (13): 921–5. doi:10.1056/NEJM199309233291304. PMID 8361506.
- ↑ Towbin JA, Ortiz-Lopez R (1994). “X-linked dilated cardiomyopathy”. N Engl J Med. 330 (5): 369–70. PMID 8123157.
- ↑ Muntoni F, Melis MA, Ganau A, Dubowitz V (1995). “Transcription of the dystrophin gene in normal tissues and in skeletal muscle of a family with X-linked dilated cardiomyopathy”. Am J Hum Genet. 56 (1): 151–7. PMC 1801315. PMID 7825571.
- ↑ Schönberger J, Levy H, Grünig E, Sangwatanaroj S, Fatkin D, MacRae C; et al. (2000). “Dilated cardiomyopathy and sensorineural hearing loss: a heritable syndrome that maps to 6q23-24”. Circulation. 101 (15): 1812–8. PMID 10769282.
- ↑ Schönberger J, Wang L, Shin JT, Kim SD, Depreux FF, Zhu H; et al. (2005). “Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss”. Nat Genet. 37 (4): 418–22. doi:10.1038/ng1527. PMID 15735644.
- ↑ Arimura T, Hayashi T, Matsumoto Y, Shibata H, Hiroi S, Nakamura T; et al. (2007). “Structural analysis of four and half LIM protein-2 in dilated cardiomyopathy”. Biochem Biophys Res Commun. 357 (1): 162–7. doi:10.1016/j.bbrc.2007.03.128. PMID 17416352.
- ↑ Murakami T, Hayashi YK, Noguchi S, Ogawa M, Nonaka I, Tanabe Y; et al. (2006). “Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness”. Ann Neurol. 60 (5): 597–602. doi:10.1002/ana.20973. PMID 17036286.
- ↑ Taylor MR, Ku L, Slavov D, Cavanaugh J, Boucek M, Zhu X; et al. (2007). “Danon disease presenting with dilated cardiomyopathy and a complex phenotype”. J Hum Genet. 52 (10): 830–5. doi:10.1007/s10038-007-0184-8. PMID 17899313.
- ↑ Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z; et al. (2003). “Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction”. J Am Coll Cardiol. 42 (11): 2014–27. PMID 14662268.
- ↑ Arimura T, Inagaki N, Hayashi T, Shichi D, Sato A, Hinohara K; et al. (2009). “Impaired binding of ZASP/Cypher with phosphoglucomutase 1 is associated with dilated cardiomyopathy”. Cardiovasc Res. 83 (1): 80–8. doi:10.1093/cvr/cvp119. PMID 19377068.
- ↑ Arimura T, Hayashi T, Terada H, Lee SY, Zhou Q, Takahashi M; et al. (2004). “A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C.” J Biol Chem. 279 (8): 6746–52. doi:10.1074/jbc.M311849200. PMID 14660611.
- ↑ 34.0 34.1 Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P; et al. (2008). “Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy”. Clin Transl Sci. 1 (1): 21–6. doi:10.1111/j.1752-8062.2008.00017.x. PMC 2633921. PMID 19412328.
- ↑ Meyer T, Ruppert V, Ackermann S, Richter A, Perrot A, Sperling SR; et al. (2013). “Novel mutations in the sarcomeric protein myopalladin in patients with dilated cardiomyopathy”. Eur J Hum Genet. 21 (3): 294–300. doi:10.1038/ejhg.2012.173. PMC 3573205. PMID 22892539.
- ↑ Carniel E, Taylor MR, Sinagra G, Di Lenarda A, Ku L, Fain PR; et al. (2005). “Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy”. Circulation. 112 (1): 54–9. doi:10.1161/CIRCULATIONAHA.104.507699. PMID 15998695.
- ↑ Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M; et al. (2002). “Novel mutations in sarcomeric protein genes in dilated cardiomyopathy”. Biochem Biophys Res Commun. 298 (1): 116–20. PMID 12379228.
- ↑ 38.0 38.1 Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B; et al. (2000). “Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy”. N Engl J Med. 343 (23): 1688–96. doi:10.1056/NEJM200012073432304. PMID 11106718.
- ↑ Hassel D, Dahme T, Erdmann J, Meder B, Huge A, Stoll M; et al. (2009). “Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy”. Nat Med. 15 (11): 1281–8. doi:10.1038/nm.2037. PMID 19881492.
- ↑ 40.0 40.1 Li D, Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S; et al. (2006). “Mutations of presenilin genes in dilated cardiomyopathy and heart failure”. Am J Hum Genet. 79 (6): 1030–9. doi:10.1086/509900. PMC 1698711. PMID 17186461.
- ↑ Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ; et al. (2009). “Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy”. J Am Coll Cardiol. 54 (10): 930–41. doi:10.1016/j.jacc.2009.05.038. PMC 2782634. PMID 19712804.
- ↑ Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M; et al. (2010). “Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy”. Clin Transl Sci. 3 (3): 90–7. doi:10.1111/j.1752-8062.2010.00198.x. PMC 2898174. PMID 20590677.
- ↑ Mann SA, Castro ML, Ohanian M, Guo G, Zodgekar P, Sheu A; et al. (2012). “R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy”. J Am Coll Cardiol. 60 (16): 1566–73. doi:10.1016/j.jacc.2012.05.050. PMID 22999724.
- ↑ Morales A, Painter T, Li R, Siegfried JD, Li D, Norton N; et al. (2010). “Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy”. Circulation. 121 (20): 2176–82. doi:10.1161/CIRCULATIONAHA.109.931220. PMC 2900861. PMID 20458009.
- ↑ Cheng J, Morales A, Siegfried JD, Li D, Norton N, Song J; et al. (2010). “SCN5A rare variants in familial dilated cardiomyopathy decrease peak sodium current depending on the common polymorphism H558R and common splice variant Q1077del”. Clin Transl Sci. 3 (6): 287–94. doi:10.1111/j.1752-8062.2010.00249.x. PMC 3026282. PMID 21167004.
- ↑ Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ; et al. (2005). “Sodium channel mutations and susceptibility to heart failure and atrial fibrillation”. JAMA. 293 (4): 447–54. doi:10.1001/jama.293.4.447. PMC 2039897. PMID 15671429.
- ↑ McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E; et al. (2004). “SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia”. Circulation. 110 (15): 2163–7. doi:10.1161/01.CIR.0000144458.58660.BB. PMID 15466643.
- ↑ D’Adamo P, Fassone L, Gedeon A, Janssen EA, Bione S, Bolhuis PA; et al. (1997). “The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies”. Am J Hum Genet. 61 (4): 862–7. doi:10.1086/514886. PMC 1715993. PMID 9382096.
- ↑ Bissler JJ, Tsoras M, Göring HH, Hug P, Chuck G, Tombragel E; et al. (2002). “Infantile dilated X-linked cardiomyopathy, G4.5 mutations, altered lipids, and ultrastructural malformations of mitochondria in heart, liver, and skeletal muscle”. Lab Invest. 82 (3): 335–44. PMID 11896212.
- ↑ Taylor MR, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR; et al. (2005). “Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy”. Hum Mutat. 26 (6): 566–74. doi:10.1002/humu.20250. PMID 16247757.
- ↑ 51.0 51.1 Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H; et al. (2004). “Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy”. J Am Coll Cardiol. 44 (10): 2033–40. doi:10.1016/j.jacc.2004.08.027. PMID 15542288.
- ↑ Murphy RT, Mogensen J, Shaw A, Kubo T, Hughes S, McKenna WJ (2004). “Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy”. Lancet. 363 (9406): 371–2. doi:10.1016/S0140-6736(04)15468-8. PMID 15070570.
- ↑ Carballo S, Robinson P, Otway R, Fatkin D, Jongbloed JD, de Jonge N; et al. (2009). “Identification and functional characterization of cardiac troponin I as a novel disease gene in autosomal dominant dilated cardiomyopathy”. Circ Res. 105 (4): 375–82. doi:10.1161/CIRCRESAHA.109.196055. PMID 19590045.
- ↑ Hanson EL, Jakobs PM, Keegan H, Coates K, Bousman S, Dienel NH; et al. (2002). “Cardiac troponin T lysine 210 deletion in a family with dilated cardiomyopathy”. J Card Fail. 8 (1): 28–32. PMID 11862580.
- ↑ Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S; et al. (2009). “Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy”. Circ Cardiovasc Genet. 2 (4): 306–13. doi:10.1161/CIRCGENETICS.108.846733. PMC 2900844. PMID 20031601.
- ↑ Sfichi-Duke L, Garcia-Cazarin ML, Sumandea CA, Sievert GA, Balke CW, Zhan DY; et al. (2010). “Cardiomyopathy-causing deletion K210 in cardiac troponin T alters phosphorylation propensity of sarcomeric proteins”. J Mol Cell Cardiol. 48 (5): 934–42. doi:10.1016/j.yjmcc.2010.01.005. PMC 2854196. PMID 20079745.
- ↑ Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, Ohta M; et al. (2002). “Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy”. Proc Natl Acad Sci U S A. 99 (2): 913–8. doi:10.1073/pnas.022628899. PMC 117405. PMID 11773635.
- ↑ Otten E, Lekanne Dit Deprez RH, Weiss MM, van Slegtenhorst M, Joosten M, van der Smagt JJ; et al. (2010). “Recurrent and founder mutations in the Netherlands: mutation p.K217del in troponin T2, causing dilated cardiomyopathy”. Neth Heart J. 18 (10): 478–85. PMC 2954300. PMID 20978592.
- ↑ Olson TM, Kishimoto NY, Whitby FG, Michels VV (2001). “Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy”. J Mol Cell Cardiol. 33 (4): 723–32. doi:10.1006/jmcc.2000.1339. PMID 11273725.
- ↑ Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S; et al. (2010). “Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy”. J Am Coll Cardiol. 55 (4): 320–9. doi:10.1016/j.jacc.2009.11.017. PMC 3000630. PMID 20117437.
- ↑ Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T; et al. (2002). “Titin mutations as the molecular basis for dilated cardiomyopathy”. Biochem Biophys Res Commun. 291 (2): 385–93. doi:10.1006/bbrc.2002.6448. PMID 11846417.
- ↑ Gerull B, Gramlich M, Atherton J, McNabb M, Trombitás K, Sasse-Klaassen S; et al. (2002). “Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy”. Nat Genet. 30 (2): 201–4. doi:10.1038/ng815. PMID 11788824.
- ↑ Siu BL, Niimura H, Osborne JA, Fatkin D, MacRae C, Solomon S; et al. (1999). “Familial dilated cardiomyopathy locus maps to chromosome 2q31”. Circulation. 99 (8): 1022–6. PMID 10051295.
- ↑ Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM (2002). “Metavinculin mutations alter actin interaction in dilated cardiomyopathy”. Circulation. 105 (4): 431–7. PMID 11815424.
- ↑ Vasile VC, Will ML, Ommen SR, Edwards WD, Olson TM, Ackerman MJ (2006). “Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy”. Mol Genet Metab. 87 (2): 169–74. doi:10.1016/j.ymgme.2005.08.006. PMID 16236538.
- ↑ Haghighi K, Chen G, Sato Y, Fan GC, He S, Kolokathis F; et al. (2008). “A human phospholamban promoter polymorphism in dilated cardiomyopathy alters transcriptional regulation by glucocorticoids”. Hum Mutat. 29 (5): 640–7. doi:10.1002/humu.20692. PMID 18241046.
- ↑ Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA; et al. (2006). “A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy”. Proc Natl Acad Sci U S A. 103 (5): 1388–93. doi:10.1073/pnas.0510519103. PMC 1360586. PMID 16432188.
- ↑ Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U; et al. (2003). “Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban”. Science. 299 (5611): 1410–3. doi:10.1126/science.1081578. PMID 12610310.
- ↑ Ha KN, Masterson LR, Hou Z, Verardi R, Walsh N, Veglia G; et al. (2011). “Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A.” Proc Natl Acad Sci U S A. 108 (7): 2735–40. doi:10.1073/pnas.1013987108. PMC 3041113. PMID 21282613.
- ↑ DeWitt MM, MacLeod HM, Soliven B, McNally EM (2006). “Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy”. J Am Coll Cardiol. 48 (7): 1396–8. doi:10.1016/j.jacc.2006.07.016. PMID 17010801.
- ↑ Posch MG, Perrot A, Geier C, Boldt LH, Schmidt G, Lehmkuhl HB; et al. (2009). “Genetic deletion of arginine 14 in phospholamban causes dilated cardiomyopathy with attenuated electrocardiographic R amplitudes”. Heart Rhythm. 6 (4): 480–6. doi:10.1016/j.hrthm.2009.01.016. PMID 19324307.
- ↑ Haghighi K, Pritchard T, Bossuyt J, Waggoner JR, Yuan Q, Fan GC; et al. (2012). “The human phospholamban Arg14-deletion mutant localizes to plasma membrane and interacts with the Na/K-ATPase”. J Mol Cell Cardiol. 52 (3): 773–82. doi:10.1016/j.yjmcc.2011.11.012. PMC 3376549. PMID 22155237.
- ↑ van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC; et al. (2012). “Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy”. Eur J Heart Fail. 14 (11): 1199–207. doi:10.1093/eurjhf/hfs119. PMC 3475434. PMID 22820313.
- ↑ Levitas A, Muhammad E, Harel G, Saada A, Caspi VC, Manor E; et al. (2010). “Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase”. Eur J Hum Genet. 18 (10): 1160–5. doi:10.1038/ejhg.2010.83. PMC 2987458. PMID 20551992.
- ↑ Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O’Cochlain F, Gao F; et al. (2004). “ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating”. Nat Genet. 36 (4): 382–7. doi:10.1038/ng1329. PMC 1995438. PMID 15034580.
- ↑ Małek LA, Labib S, Mazurkiewicz L, Saj M, Płoski R, Tesson F; et al. (2011). “A new c.1621 C > G, p.R541G lamin A/C mutation in a family with DCM and regional wall motion abnormalities (akinesis/dyskinesis): genotype-phenotype correlation”. J Hum Genet. 56 (1): 83–6. doi:10.1038/jhg.2010.137. PMID 21085127.
- ↑ Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M; et al. (1999). “Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease”. N Engl J Med. 341 (23): 1715–24. doi:10.1056/NEJM199912023412302. PMID 10580070.
- ↑ Sébillon P, Bouchier C, Bidot LD, Bonne G, Ahamed K, Charron P; et al. (2003). “Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations”. J Med Genet. 40 (8): 560–7. PMC 1735561. PMID 12920062.
- ↑ van der Kooi AJ, Bonne G, Eymard B, Duboc D, Talim B, Van der Valk M; et al. (2002). “Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy”. Neurology. 59 (4): 620–3. PMID 12196663.
- ↑ Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J (2013). “Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics”. Nature. 497 (7450): 507–11. doi:10.1038/nature12105. PMC 3666313. PMID 23644458.
- ↑ Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E; et al. (2003). “Natural history of dilated cardiomyopathy due to lamin A/C gene mutations”. J Am Coll Cardiol. 41 (5): 771–80. PMID 12628721.
- ↑ Charniot JC, Pascal C, Bouchier C, Sébillon P, Salama J, Duboscq-Bidot L; et al. (2003). “Functional consequences of an LMNA mutation associated with a new cardiac and non-cardiac phenotype”. Hum Mutat. 21 (5): 473–81. doi:10.1002/humu.10170. PMID 12673789.
- ↑ Brodsky GL, Muntoni F, Miocic S, Sinagra G, Sewry C, Mestroni L (2000). “Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement”. Circulation. 101 (5): 473–6. PMID 10662742.
- ↑ Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A; et al. (2007). “Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity”. Hum Mol Genet. 16 (23): 2816–33. doi:10.1093/hmg/ddm238. PMID 17761684.
- ↑ Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM (2012). “Population-based variation in cardiomyopathy genes”. Circ Cardiovasc Genet. 5 (4): 391–9. doi:10.1161/CIRCGENETICS.112.962928. PMC 3495587. PMID 22763267.
- ↑ Jefferies JL, Towbin JA (2010). “Dilated cardiomyopathy”. Lancet. 375 (9716): 752–62. doi:10.1016/S0140-6736(09)62023-7. PMID 20189027.
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Sachin Shah, M.D. Ogheneochuko Ajari, MB.BS, MS [2]
Overview
There are many causes of dilated cardiomyopathy. The most common cause is idiopathic in 50% of cases. The next most common cause is myocarditis which is responsible for 10% of cases. The high percentage of idiopathic dilated cardiomyopathy may be related to the difficulty in diagnosing viral myocarditis. Other common causes include substance abuse, connective tissue disease, pregnancy, medications, nutritional deficiencies, infiltrative diseases and toxins. There are varying degrees of severity of the disease. Some forms are reversible and some are irreversible; some patients may be completely asymptomatic and some may require cardiac transplantation.
Causes
There are several recorded causes for dilated cardiomyopathy. [1][2][3][4][5]
Life Threatening Causes
Life-threatening causes include conditions which may result in death or permanent disability within 24 hours if left untreated.
Common Causes
- Antiretroviral drugs
- Chemotherapeutic agents (such as Doxorubicin)
- Connective tissue disease
- Eosinophilic cardiomyopathy
- Hemochromatosis
- HIV infection
- Hypertensive heart disease
- Idiopathic
- Ischemic cardiomyopathy
- Myocarditis
- Nutritional deficiencies (such as thiamine or selenium)
- Peripartum cardiomyopathy
- Sarcoidosis
- Sleep apnea
- Substance abuse (alcohol abuse or cocaine abuse)
- Toxins (such as cobalt, lead or beryllium)
Causes by Organ System
Causes in Alphabetical Order
References
- ↑ Felker GM, Thompson RE, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med 2000 Apr 13;342(14):1077-84.
- ↑ Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK (2016) The Diagnosis and Evaluation of Dilated Cardiomyopathy. J Am Coll Cardiol 67 (25):2996-3010. DOI:10.1016/j.jacc.2016.03.590 PMID: 27339497
- ↑ Baris L, Cornette J, Johnson MR, Sliwa K, Roos-Hesselink JW (2019). “Peripartum cardiomyopathy: disease or syndrome?”. Heart. 105 (5): 357–362. doi:10.1136/heartjnl-2018-314252. PMC 6613742 Check
|pmc=value (help). PMID 31693481. - ↑ Weintraub RG, Semsarian C, Macdonald P (2017). “Dilated cardiomyopathy”. Lancet. 390 (10092): 400–414. doi:10.1016/S0140-6736(16)31713-5. PMID 28190577.
- ↑ Favalli V, Serio A, Grasso M, Arbustini E (2016). “Genetic causes of dilated cardiomyopathy”. Heart. 102 (24): 2004–2014. doi:10.1136/heartjnl-2015-308190. PMID 27634407.
Differentiating Dilated cardiomyopathy from other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Abdelrahman Ibrahim Abushouk, MD[2]
Overview
Dilated cardiomyopathy should be differentiated from other causes of cardiac dysfunction, in particular acute coronary syndrome, other cardiomyopathies (hypertrophic, restrictive, and ARVC/D), myocarditis, pericarditis, and cardiac toxicities.
Differentiating Dilated Cardiomyopathy from other Diseases
Dilated cardiomyopathy should be differentiated from other causes of cardiac dysfunction, in particular acute coronary syndrome, other cardiomyopathies (hypertrophic, restrictive, and ARVC/D), myocarditis, pericarditis, and cardiac toxicities.[1][2][3][4]
| Disorders | Etiology | Clinical Presentation | Laboratory Findings | Electrocardiogram | Echocardiography |
|---|---|---|---|---|---|
| Dilated Cardiomyopathy |
|
|
|
|
|
| Acute Coronary Syndrome |
|
|
| ||
| Acute Pericarditis |
|
|
|
||
| Amphetamine/Cocaine Cardiomyopathy |
|
|
|
|
|
| Arrhythmogenic right ventricular
cardiomyopathy (ARVC/D) |
|
|
Diagnostic criteria are based on:
|
|
|
| Wet Beriberi |
|
|
|
In advanced beriberi, heart failure occurs.
|
In advanced beriberi, heart failure occurs.
|
| Cardiac Tamponade |
|
|
|
| |
| Hyperthyroidism |
|
|
|
|
The following may be present:
|
| Hypertrophic Cardiomyopathy |
|
|
|
| |
| Left ventricular noncompaction |
|
|
|
|
|
| Myocarditis |
|
|
|
| |
| Restrictive Cardiomyopathy | Systemic diseases, such as |
|
|
|
|
References
- ↑ Amosova EN (1992). “[Differential diagnosis of dilated cardiomyopathy]”. Klin Med (Mosk). 70 (3–4): 14–9. PMID 1507837.
- ↑ Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE; et al. (2019). “Dilated cardiomyopathy”. Nat Rev Dis Primers. 5 (1): 32. doi:10.1038/s41572-019-0084-1. PMID 31073128.
- ↑ Gurevich MA, Gordienko BV (2003). “[Dilated and ischemic cardiomyopathy: differential diagnosis]”. Klin Med (Mosk). 81 (9): 68–71. PMID 14598597.
- ↑ Gurevich MA, Gordienko BV (2003). “[Dilated and ischemic cardiomyopathy: differential diagnosis]”. Klin Med (Mosk). 81 (9): 68–71. PMID 14598597.
Epidemiology and Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Abdelrahman Ibrahim Abushouk, MD[2] Sachin Shah, M.D.
Overview
The prevalence of dilated cardiomyopathy is approximately 36 per 100,000 individuals worldwide. It has a high mortality rate of up to 50%. Dilated cardiomyopathy is most likely to occur between the ages of 20-60, is three times as likely to occur in males over females, and is 2.5 times more likely to occur in African Americans.
Epidemiology and Demographics
Incidence
- The incidence of dilated cardiomyopathy is approximately 4.5 per 100,000 individuals per year worldwide.
Prevalence
- The prevalence of dilated cardiomyopathy is approximately 36 per 100,000 individuals worldwide.
Case-fatality rate/Mortality rate
- The mortality rate of dilated cardiomyopathy is quite high (up to 50%).
Age
- Dilated cardiomyopathy can occur at any age (although it is more likely between the ages of 20-60).[1]
Gender
- In dilated cardiomyopathy, there is a male predominance (3:1 male:female).[2]
Race
- Dilated cardiomyopathy is 2.5 times more likely to occur in African Americans.[3].
References
- ↑ Dec GW, Fuster V. Idiopathic Dilated Cardiomyopathy. N Engl J Med 1994 Dec 8;331(23):1564-75. PMID 7969328
- ↑ Robbins Basic Pathology, 7th edition. Kumar, Cotran, Robbins. ISBN 0-7216-9274-5
- ↑ Coughlin SS, Labenberg JR, Tefft MC. Black-white differences in idiopathic dilated cardiomyopathy: the Washington DC Dilated Cardiomyopathy Study. Epidemiology. 1993;4:165-72. PMID 8452906
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Abdelrahman Ibrahim Abushouk, MD[2]
Overview
Common risk factors in the development of dilated cardiomyopathy include genetic inheritance, nutritional deficiencies, substance abuse, occupational exposure to toxins, and viral infections.
Risk Factors
Common risk factors in the development of dilated cardiomyopathy include genetic inheritance, nutritional deficiencies, substance abuse, occupational exposure to toxins, and viral infections.[1][2][3][4]
Common Risk Factors
- Common risk factors in the development of dilated cardiomyopathy include:
- Genetic inheritance
- Substance (cocaine/amphetamine) and alcohol abuse
- Drugs (Doxorubicin, Cyclophosphamide, and antiretroviral drugs)
- Viral infections as adenovirus and coxsackie virus.
- Occupational exposure to industrial toxins as cobalt, lead or beryllium
- Nutritional deficiencies as thiamine and selenium.
Less Common Risk Factors
- Less common risk factors in the development of dilated cardiomyopathy include:
References
- ↑ Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL; et al. (2000). “Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy”. N Engl J Med. 342 (15): 1077–84. doi:10.1056/NEJM200004133421502. PMID 10760308.
- ↑ McNally EM, Mestroni L (2017). “Dilated Cardiomyopathy: Genetic Determinants and Mechanisms”. Circ Res. 121 (7): 731–748. doi:10.1161/CIRCRESAHA.116.309396. PMC 5626020. PMID 28912180.
- ↑ Lipshultz SE (1998). “Dilated cardiomyopathy in HIV-infected patients”. N Engl J Med. 339 (16): 1153–5. doi:10.1056/NEJM199810153391609. PMID 9770563.
- ↑ Li X, Nie Y, Lian H, Hu S (2018). “Histopathologic features of alcoholic cardiomyopathy compared with idiopathic dilated cardiomyopathy”. Medicine (Baltimore). 97 (39): e12259. doi:10.1097/MD.0000000000012259. PMC 6181549. PMID 30278496.
Screening
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Abdelrahman Ibrahim Abushouk, MD[2]
Overview
The current guidelines recommend screening for dilated cardiomyopathy in individuals with 2 or 3 family members with primary dilated cardiomyopathy. Screening can be performed using electrocardiograms and echocardiography to measure the size and function of the left ventricle. An underlying genetic mutation in the 40 genes (currently assessed in FCD genetic testing) can be detected in 30 to 40% of FCD patients.
Screening
Screening is only recommended in individuals who have a family history of familial dilated cardiomyopathy (FDC).
- In case of 2 or 3 family members affected with primary DCM, screening of first degree relatives, using electrocardiogram and echocardiography allows the identification of FDC according to guidelines from the Cardiac Genetic Diseases Council.
- FDC can be identified in 20–35% of DCM cases, while the remaining are classified as ‘idiopathic’.[1]
- FCD is primarily an autosomal dominant disease; however, cases with autosomal recessive or X-linked inheritance have been recognized.
- Genetic studies have revealed an underlying genetic mutation in 30 to 40% of patients with FCD.[2]
- About 40 genes are currently included in genetic testing; however, mutations in >60 other genes have been linked to FCD, but are yet to be included.
- A cohort study by Haas et al. showed that the genetic mutations in FCD overlap with other cardiomyopathies, highlighting the value of genetic testing in different types of cardiomyopathy.[3]
- Current studies are focusing on the role of multiple mutations, enhancer region mutations, copy number variation, and intronic variants.[4]
References
- ↑ Fatkin D, members of the CSANZ Cardiac Genetic Diseases Council Writing Group (2011). “Guidelines for the diagnosis and management of familial dilated cardiomyopathy”. Heart Lung Circ. 20 (11): 691–3. doi:10.1016/j.hlc.2011.07.008. PMID 21885340.
- ↑ McNally EM, Golbus JR, Puckelwartz MJ (2013). “Genetic mutations and mechanisms in dilated cardiomyopathy”. J Clin Invest. 123 (1): 19–26. doi:10.1172/JCI62862. PMC 3533274. PMID 23281406.
- ↑ Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R; et al. (2015). “Atlas of the clinical genetics of human dilated cardiomyopathy”. Eur Heart J. 36 (18): 1123–35a. doi:10.1093/eurheartj/ehu301. PMID 25163546.
- ↑ Sweet M, Taylor MR, Mestroni L (2015). “Diagnosis, prevalence, and screening of familial dilated cardiomyopathy”. Expert Opin Orphan Drugs. 3 (8): 869–876. doi:10.1517/21678707.2015.1057498. PMC 4988677. PMID 27547593.
Natural History, Complications and Prognosis
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]Associate Editor(s)-in-Chief: Abdelrahman Ibrahim Abushouk, MD[2]
Overview
The natural history of dilated cardiomyopathy has significantly improved following the advances in medical therapy and introduction of cardiac resynchronization and implantable defibrillators. However, patients with DCM are still prone to complications as heart failure, arrhythmias, arterial embolism and sudden cardiac death. There are several prognostic indicators when evaluating dilated cardiomyopathy, the most important one being ejection fraction. Complications as a result of dilated cardiomyopathy include heart failure, aortic and mitral valve regurgitation, emboli, edema, arrhythmias and sudden cardiac arrest.
Natural History, Complications, and Prognosis
Natural History
- Dilated cardiomyopathy is the final common pathway for different etiologic mechanisms.
- During the initial visit of a patient with DCM, the clinician should in fact consider all the potentially reversible causes of left ventricular dysfunction, likely to benefit from specific therapeutic intervention.
- The onset of DCM can be at times indistinguishable from other conditions which can be specifically recovered by correcting the underlying problem.[1]
- The natural history of DCM has thus significantly changed in the last few years, particularly after the introduction and utilization of ACE-inhibitors, betablockers, anti-aldosterone drugs and nonpharmacological treatments, such as cardiac resynchronization (CRT) and implantable defibrillator (ICD).[2]
- However, still about 2% of patients with DCM died of sudden cardiac death after the diagnosis and the risk of other complications as heart failure, arterial infarctions, and valvular insufficiency remains high.[3]
Complications
The following complications may occur in patients with dilated cardiomyopathy.[3]
- Heart failure: may occur as the end result of DCM pathogenesis.
- Heart valve regurgitation
- Arrhythmias: as conduction delays, bundle branch blocks, and tachyarrhythmias.
- Edema: Central (pulmonary congestion and hepatomegaly) and peripheral (pedal).
- Embolism: causing arterial infarcts as renal infarcts and stroke.
- Sudden cardiac arrest: may occur in 2% of patients with DCM.
-
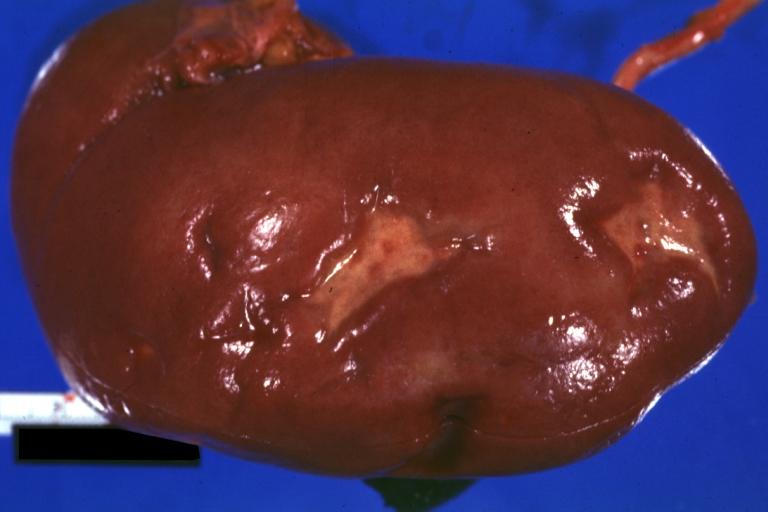 Kidney: Infarct Remote: Gross external view with capsule removed two old and very typical infarct scars 27 year old person with dilated cardiomyopathy
Kidney: Infarct Remote: Gross external view with capsule removed two old and very typical infarct scars 27 year old person with dilated cardiomyopathy -
 Brain: Infarct: Healing large MCA and PICA probably embolic 64 year old female chronic obstructive pulmonary disease and cardiomyopathy with atrial fibrillation
Brain: Infarct: Healing large MCA and PICA probably embolic 64 year old female chronic obstructive pulmonary disease and cardiomyopathy with atrial fibrillation


Prognosis
There are many prognostic factors which can be evaluated in a patient with dilated cardiomyopathy. The most important prognostic indicator is a decreased ejection fraction, in addition increased left ventricular size and right ventricular dilation are independent indicators of a poor prognosis. As is in most types of heart failure a poor NYHA functional class and increased PASP (>35mmHg) are also poor prognostic indicators. Other findings that infer a poor prognosis are as follows: Maximal O2 uptake of < 12mL/kg / minute on exercise testing, LBBB (left bundle branch block), non sustained ventricular tachycardia, syncope, hyponatremia with a serum sodium less than 135, elevated norepinephrine, ANP (atrial natriuretic peptide) and renin levels (not routinely measured in clinical practice), elevated PCWP (pulmonary capillary wedge pressure) > 18mmHg and low cardiac index < 2.5L/min/m^2.[4][5]
References
- ↑ Di Lenarda A, Pinamonti B, Mestroni L, Salvi A, Sabbadini G, Gregori D; et al. (2004). “[How the natural history of dilated cardiomyopathy has changed. Review of the Registry of Myocardial Diseases of Trieste]”. Ital Heart J Suppl. 5 (4): 253–66. PMID 15185463.
- ↑ Merlo M, Gentile P, Naso P, Sinagra G (2017). “The natural history of dilated cardiomyopathy: how has it changed?”. J Cardiovasc Med (Hagerstown). 18 Suppl 1: e161–e165. doi:10.2459/JCM.0000000000000459. PMID 27828827.
- ↑ 3.0 3.1 Zecchin M, Merlo M, Pivetta A, Barbati G, Lutman C, Gregori D; et al. (2012). “How can optimization of medical treatment avoid unnecessary implantable cardioverter-defibrillator implantations in patients with idiopathic dilated cardiomyopathy presenting with “SCD-HeFT criteria?““. Am J Cardiol. 109 (5): 729–35. doi:10.1016/j.amjcard.2011.10.033. PMID 22176998.
- ↑ Castelli G, Fornaro A, Ciaccheri M, Dolara A, Troiani V, Tomberli B; et al. (2013). “Improving survival rates of patients with idiopathic dilated cardiomyopathy in Tuscany over 3 decades: impact of evidence-based management”. Circ Heart Fail. 6 (5): 913–21. doi:10.1161/CIRCHEARTFAILURE.112.000120. PMID 23888044.
- ↑ Hagar A, Pu XB, Chen SJ, Shah JP, Chen M (2019). “Clinical characteristics, treatment and prognosis of patients with idiopathic dilated cardiomyopathy: a tertiary center experience”. J Geriatr Cardiol. 16 (4): 320–328. doi:10.11909/j.issn.1671-5411.2019.04.004. PMC 6503477 Check
|pmc=value (help). PMID 31105752.
Diagnosis
Diagnosis
History and Symptoms | Physical Examination | Laboratory Findings | Electrocardiogram | Chest X-Ray | CT | MRI | Echocardiography | Other Imaging Findings | Genetic testing | Other Diagnostic Studies
Treatment
Treatment
Medical Therapy | Surgery | Primary Prevention | Secondary Prevention | Cost-Effectiveness of Therapy | Future or Investigational Therapies
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH