
Hypertrophic cardiomyopathy
For patient information click here
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Soroush Seifirad, M.D.[2]
Synonyms and keywords: Hypertrophic cardiomyopathy or HCM, Asymmetric septal hypertrophy or ASH, Hypertrophic obstructive cardiomyopathy, HOCM, Idiopathic hypertrophic subaortic stenosis or IHSS, familial isolated hypertrophic obstructive cardiomyopathy, familial isolated hypertrophic subaortic stenosis, familial hypertrophic subaortic stenosis, idiopathic hypertrophic subaortic stenosis, familial hypertrophic obstructive cardiomyopathy, idiopathic hypertrophic obstructive cardiomyopathy, primitive hypertrophic obstructive cardiomyopathy, primitive hypertrophic subaortic stenosis, muscular subaortic stenosis, apical hypertrophic cardiomyopathy, which is also known as nonobstructive hypertrophic cardiomyopathy and Japanese variant hypertrophic cardiomyopathy or Yamaguchi syndrome.
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Soroush Seifirad, M.D.[2]
Overview
Hypertrophic cardiomyopathy, or HCM, is a disease of the myocardium in which a portion of the myocardium is hypertrophied without any alternate known cause such as hypertension, amyloid or aortic stenosis. Although HCM has gained notoriety as a leading cause of sudden cardiac death in young athletes, it should be noted that HCM is a cause of sudden cardiac death in any age group and may be associated with cardiac morbidity and disabling cardiac symptoms as well. There are two variants of hypertrophic cardiomyopathy: an obstructive variant, and a non-obstructive variant. A non-obstructive variant of HCM is known as apical hypertrophic cardiomyopathy ,which is also known as nonobstructive hypertrophic cardiomyopathy and Japanese variant hypertrophic cardiomyopathy or the Yamaguchi variant (since the first cases described were all in individuals of Japanese descent). In hypertrophic cardiomyopathy (HCM), the sarcomeres in the heart replicate causing heart muscle cells to increase in size, which results in the thickening of the heart muscle. In addition, the normal alignment of muscle cells is disrupted, a phenomenon known as myocardial disarray. Myosin heavy chain mutations are associated with the development of familial hypertrophic cardiomyopathy. HCM also causes disruptions of the electrical functions of the heart. Hypertrophic cardiomyopathy is most commonly due to a mutation in one of 14 sarcomeric genes that results in a mutated protein in the sarcomere, the primary component of the myocyte. While most literature so far focuses on European, American, and Japanese populations, HCM appears in all racial groups. The prevalence of HCM is about 0.2% to 0.5% of the general population. HCM is frequently asymptomatic until sudden cardiac death, and that is why some experts suggest routinely screening certain populations for this disease. Echocardiographuc surveys in general population showed HCM in approximately 1 in every 500 people (0.2% of the general population). Only 15% of HCM patients have been diagnosed, which means the majority of patients with HCM are undiagnosed. The symptoms associated with hypertrophic cardiomyopathy are quite variable and range from no symptoms, to the development of heart failure, or sudden cardiac death. The symptoms may vary tremendously from individual even within a family (different penetrance). The timing of symptom onset is quite variable as well and may range from infancy to adulthood. Symptoms may include chest pain, dizziness, fainting, especially during exercise, heart failure (in some patients), hypertension, dizziness during activity or when standing up suddenly, sensation of feeling the heart beat (palpitations), shortness of breath, fatigue, reduced activity tolerance, and shortness of breath when lying down (orthopnea). The medical management of the patient with hypertrophic cardiomyopathy involves minimizing diastolic dysfunction, reducing left ventricular outflow tract obstruction, optimizing heart failure management, maintaining normal sinus rhythm, rate control and anticoagulation in the presence of atrial fibrillation, and implantation of an automatic implantable cardiac defibrillator in selected patients such as those who survive sudden cardiac death.
Historical Perspective
The first case of hypertrophic cardiomyopathy (HCM) was described by in 1869 Henri Liouville in the Gazette Medecine Paris. In 1907 Dr. A. Schmincke, a German pathologist, described two hearts with left ventricular hypertrophy; both came from women in their mid-fifties. Levy and von Glahn in 1944, from Colombia University in New York, published a series of cases which resembles HCM. In 1949, William Evans, a London cardiologist, described familial occurrence of cardiac hypertrophy in a series of patients which were similar to those described in the paper by Levy and von Glahn. In 1961 Paré et al. reported thirty members of five generations of a French Canadian family in Quebec in whom the condition was inherited in an autosomal dominant manner. In 1958 Teare, an English pathologist, described eight cases of asymmetric cardiac muscle hypertrophy, he thought that they might be benign cardiac tumors. Seven of these caused sudden death in young adults. Teare named the condition “Asymmetrical Hypertrophy of the Heart.” In 1959 Sir Russell Brock described a young man with angina and a subaortic stenosis and a subaortic intraventricular pressure gradient. Morrow and Braunwald published their first report in the same year, followed by several other reports. The sudden cardiac deaths of 387 young American athletes (under age 35) were analyzed in a 2003 medical review, and HCM was the leading cause of sudden cardiac death in athletes. In 1961, Morrow described a surgical procedure to relieve the obstruction, which is still the most widely used method of surgical treatment. In 1962, with respect to the observed intensification of obstruction in HCM with the beta-adrenergic agonists, Braunwald suggested the use of newly developed beta-blockers. In 1964, Braunwald reported beta-blockers beneficial hemodynamic effects. 1967, the clinical benefits of treatment with beta blockers in patients with HCM has been proved to the scientific society.
Classification
There are two variants of hypertrophic cardiomyopathy: an obstructive variant, and a non-obstructive variant. About 25% of individuals with hypertrophic cardiomyopathy (HCM) demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In other individuals, obstruction only occurs under certain conditions. This is known as dynamic outflow obstruction because the degree of obstruction is variable and is dependent on the amount of blood in the ventricle immediately before ventricle systole. If left ventricular outflow obstruction is present, then this syndrome has been known as wide variety of terms including: hypertrophic cardiomyopathy or HCM, asymmetric septal hypertrophy or ASH, hypertrophic obstructive cardiomyopathy, HOCM, idiopathic hypertrophic subaortic stenosis or IHSS.
A non-obstructive variant of HCM is known as apical hypertrophic cardiomyopathy, which is also known as nonobstructive hypertrophic cardiomyopathy and Japanese variant hypertrophic cardiomyopathy or the Yamaguchi variant (since the first cases described were all in individuals of Japanese descent), also known as apical hypertrophic cardiomyopathy (ApHCM) or Yamaguchi syndrome.
Pathophysiology
The progression to hypertrophic cardiomyopathy usually involves the mutations in contractile sarcomeric proteins of myocardium, which describe the presence of left ventricular hypertrophy (LVH) in the absence of an increased external load (unexplained LVH). Additionally, HCM hypertrophy is generally asymmetric.
HCM is the most common genetically transmitted cardiovascular disease. Hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins. Penetrance of HCM is incomplete, variable and time or age-related. The disease may be sporadic but affected family members are discovered in 13% of cases. More than 200 mutations involving at least 10 chromosomes encoding structural proteins of the myocyte have been discovered. These mutations have varying degrees of penetrance and even the same mutation may have variable expression, implying the superimposed effects of other genes or environmental influences. Children of a patient with HCM have a 50% chance of inheriting the trait.
Depending on the degree of obstruction of the outflow of blood from the left ventricle of the heart, HCM can be defined as obstructive or non-obstructive. About 25% of individuals with HCM demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In other individuals, obstruction only occurs under certain conditions. This is known as dynamic outflow obstruction because the degree of obstruction is variable and is dependent on the amount of blood in the ventricle immediately before ventricle systole (contraction).
Although there may be structural or functional obstruction of the left ventricular outflow tract, symptoms may arise more often from diastolic dysfunction.There is extensive periarteriolar fibrosis that results in microvascular dysfunction and impairment in coronary flow reserve in patients with hypertrophic obstructive cardiomyopathy. Individuals with HCM have some degree of left ventricular hypertrophy. In approximately 2/3rds of cases this is asymmetric hypertrophy, involving the interventricular septum, and is known as asymmetric septal hypertrophy (ASH). This is in contrast to the symmetric and concentric hypertrophy seen in aortic stenosis or hypertension. On histopathologic examination, hypertrophic cardiomyopathy is characterized by both myocardial disarrays and by periarteriolar fibrosis. Myocardial disarray can be associated with aberrant impulse conduction and arrhythmias, and periarteriolar fibrosis can be associated with myocardial ischemia.
Causes
Hypertrophic cardiomyopathy is a condition that is most often passed down through families (inherited). It is thought to result from gene mutations that control heart muscle growth. Genes involved in the pathogenesis of hypertrophic cardiomyopathy include MYH7, TNNT2, TPM1. Nevertheless, number of chronic medical conditions might be contributed to hypertrophic cardiomyopathy development, among them are thyroid disease, diabetes, and obesity, and hypertension.
Differentiating Hypertrophic Cardiomyopathy from Other Diseases
Cardiomyopathy must be differentiated from athlete heart (which is often confused with HCM on echocardiography), hypertrophy due to hypertension or aortic stenosis; as these have common clinical features, including thickened myocardium on imaging and high QRS voltage on EKGs. On the basis increased LV to aortic gradient, hypertrophic cardiomyopathy must be differentiated from sever volume depletion, subaortic stenosis, and valvular aortic stenosis.
Epidemiology and Demographics
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease. Prevalence rates have been reported between 1:500 (0.2%) and 1:3,000 (0.03%) because of variations in study designs and cohort characteristics including different age groups and ethnicity. According to the CARDIA (Coronary Artery Risk Development in Young Adults) cohort study that used standard echocardiography in 4,111 unrelated people 23 to 35 years of age, HCM prevalence is reported as 1 in 500 persons (0.2%). Nevertheless, lower prevalence has been reported in some European countries such as Germany (0.07%). Patients of all age groups may develop hypertrophic cardiomyopathy. Prevalence increased with advancing age and showed a constant yearly rise but sudden death is more prevalent in young patients, particularly athletes. The case-fatality rate is 6 per 10,000 per year in young people without symptoms of hypertrophic cardiomyopathy but in syptomatic patients a case-fatality rate is 420 and 110 deaths per 10,000 per year in tertiary referral centers and general hospital clinics respectively. Hypertrophic cardiomyopathy affects men and women equally. However, despite more frequent outflow obstruction, women with HCM are underrecognized and referred to centers later than men, often with more advanced heart failure. Greater awareness of HCM in women should lead to earlier diagnosis and treatment, with implications for improved quality of life. HCM is less prevalent in African Americans, but they are more pron to early presentation, developing heart failure, and sudden death is more prevalent due to less awareness and screening in this population.
Risk Factors
Obstructive hypertrophic cardiomyopathy (HCOM) is known as a familial genetic disorder. The most potent risk factor in the development of hypertrophic cardiomyopathy are genetic mutations in Beta-myosin heavy chain, Myosin binding protein C, and Cardiac troponin T. Genes involved in the pathogenesis of hypertrophic cardiomyopathy include but not limited to MYH7, TNNT2, TPM1. However, hypertension, thyroid disease, diabetes, and obesity also play a role in non obstructive forms of hypertrophic cardiomyopathy. This is in response to chronic effects of abnormal pressure and volumes on the heart muscle and is different from apical hypertrophy (Yamaguchi syndrome).
Screening
Genetic testing is the diagnostic study of choice to definitively diagnose hypertrophic cardiomyopathy. While definitive, these techniques can be expensive and can be difficult to access. If the mutation has already been identified in other family members, it is fairly efficient to test for that isolated mutation. Once HCM has been identified in a family, immediate testing of all family members will help to identify those at risk.
Natural History, Complications, and Prognosis
The natural history of hypertrophic cardiomyopathy is quite variable. Signs and symptoms range from none, to atrial fibrillation, to heart failure, to embolic stroke, to sudden cardiac death. Signs and symptoms are quite variable from individual to individual but are also quite variable within a given family (all of whom carry the same mutation). The disease is quite variable in the timing of its appearance and may appear anywhere from infancy to late in adult life. About 25% of HCM patients achieve normal longevity. The myosin binding proteins C genetic variant carries a good prognosis. The presence of VT / VF carries the poorest prognosis. The severity of the outflow gradient is also related to prognosis. Athletes should be screened for HOCM based upon a family history of sudden cardiac death and a murmur on physical examination. Electrocardiograms and echocardiograms are not cost effective screening tools in this population with a low pre-test probability of disease.
Diagnosis
Diagnostic Study of Choice
There is no single study of choice in the diagnosis and management of patients with HCM. Hypertrophic cardiomyopathy can be diagnosed based on clinical examination, imaging, ECG, and genetic testing. In fact, a series of studies are indicated the time of diagnosing HCM among them are Echocardiography and ECG. Echocardiography is the imaging study of choice for the diagnosis of hypertrophic cardiomyopathy. However, MRI might detect HCM sooner, and as mentioned above genetic tests are also helpful.
History and Symptoms
A large number of the patients with hypertrophic cardiomyopathy are asymptomatic or complain of mild nonspecific symptoms, Patients are often diagnosed by family screening, incidental murmur auscultation during routine examination or screening for school athletic events, or via an abnormal ECG. Nevertheless, in symptomatic patients, left ventricular outflow tract gradients and result in symptoms of dyspnea, fatigue, chest pain, and syncope are the most common presentations. The symptoms associated with hypertrophic cardiomyopathy are quite variable and range from no symptoms, to the development of heart failure, to the occurrence of sudden cardiac death. The symptoms may vary tremendously from individual even within a family. The timing of symptom onset is quite variable as well and may range from infancy to adulthood.
Physical Examination
There are numerous teachers on physical examination that allow one to distinguish hypertrophic cardiomyopathy from other conditions such as aortic stenosis. On physical examination, (as shown in the table below) maneuvers that decrease left ventricular filling augment the murmur and maneuvers that increase afterload or filling decrease the murmur.
Laboratory Findings
Genetic studies may be used in the diagnosis and screening of patients and families with known hypertrophic cardiomyopathy (HCOM). Laboratory findings consistent with the diagnosis of hypertrophic cardiomyopathy may include but not limited to mutations in the genes involved in Beta-myosin heavy chain, Myosin binding protein C, and cardiac troponin T. Genes involved in the pathogenesis of hypertrophic cardiomyopathy include MYH7, TNNT2, and TPM1.
Electrocardiogram
A 12 lead EKG is strongly recommended at the time of the initial diagnosis of hypertrophic cardiomyopathy. Common findings on an EKG in these patients include tall R waves, deep Q waves, inverted T waves, ST segment abnormalities and ‘strain pattern’ in the chest leads. The deep Q waves indicate septal hypertrophy and similarly deeply inverted T waves indicate apical hypertrophy.
X-ray
There are no x-ray findings associated with hypertrophic cardiomyopathy.
Echocardiography and Ultrasound
Echocardiography is the imaging modality of choice in the diagnosis of hypertrophic cardiomyopathy. Classically there is a small left ventricular cavity with hypertrophy out of proportion to any underlying condition that would cause LVH. The hypertrophy is often asymmetric.
CT scan
There are no CT scan findings associated with hypertrophic cardiomyopathy. However, a CT angiography may be helpful in the diagnosis of concomitant CAD in patients with hypertrophic cardiomyopathy.
MRI
Late myocardial enhancement has been associated with myocardial fibrosis and may allow for earlier detection of hypertrophic cardiomyopathy (HCM) than is currently available with echocardiography and ECG. MRI is helpful in visualizing the asymmetric thickening of the interventricular septum in patients with HCM. However, it may be more helpful than other forms of imaging to differentiate the variant types of hypertrophic cardiomyopathy. MRIcan be helpful in evaluating the extent of systolic anterior motion of the mitral valve. MRI can help visualize turbulence in the left ventricular outflow tract created by an obstruction in patients with obstructive hypertrophic cardiomyopathy.
Other Imaging Findings
Positron Emission Tomography (PET) may be helpful in the diagnosis of ischemia in patients with hypertrophic cardiomyopathy. PET studies have demonstrated that coronary flow reserve is reduced in patients with HCM. Those patients who subsequently died had a greater reduction in coronary flow reserve at baseline. It has been hypothesized that this ischemia may mediate in part the higher risk in sudden cardiac death.
Other Diagnostic Studies
Left heart catheterization can be a useful diagnostic study to ascertain the severity of the dynamic outflow obstruction and its location. Among patients who have chest discomfort or an anginal equivalent, coronary angiography carries a class I recommendation to evaluate for the presence of obstructive coronary artery disease.The prognostic value of electrophysiologic testing of patients with HOCM in the absence of spontaneous, sustained ventricular tachycardia is limited, and in fact, the study itself may be dangerous. Paced electrogram fractionation in hypertrophic cardiomyopathy may helpful in determining which patients are at risk for ventricular fibrillation.
Treatment
Medical Therapy
The medical management of the patient with hypertrophic cardiomyopathy involves minimizing diastolic dysfunction, reducing left ventricular outflow tract obstruction, optimizing heart failure management, maintaining normal sinus rhythm, rate control and anticoagulation in the presence of atrial fibrillation, and implantation of an automatic implantable cardiac defibrillator in those patients who survive sudden cardiac death.
One of the fundamental goals of treatment is to relieve disabling dyspnea and improve exercise tolerance. It should be noted that the majority of patients do not have outflow tract obstruction, and therefore would not benefit from surgery. Medical therapy is, therefore, a mainstay of treatment. Given the limited number of patients with the condition, there are few randomized trials comparing strategies/agents in the management of HCM.
In all patients with hypertrophic cardiomyopathy risk stratification is essential to attempt to ascertain which patients are at risk for sudden cardiac death. In those patients deemed to be at high risk the benefits and infrequent complications of defibrillator therapy are discussed; devices have been implanted in as many as 15% of patients at HOCM centers. Treatment symptoms of obstructive HOCM is directed towards decreasing the left ventricular outflow tract gradient and symptoms of dyspnea, chest pain and syncope.
Interventions
Alcohol septal ablation, ventricular pacing, automatic implantable cardiac defibrillator placement are among interventions used to manage patients with hypertrophic cardiomyopathy.
Surgery
Septal myectomy is a surgical treatment for hypertrophic cardiomyopathy (HCM). Septal myectomies have been successfully performed for more than 25 years.
Cardiac transplantation can be performed in patients with HOCM and has been associated with better post-operative survival than those patients transplanted for ischemic cardiomyopathy.
Primary Prevention
There is no primary prevention for hypertrophic cardiomyopathy. This is a genetic familial disorder. But there are important approaches to decrease and prevent development of sudden death and heart attack in known cases of HCM (tertiary prevention). Any activity, drug or circumstance that increases left ventricular outflow obstruction, reduced left ventricular filling, or increases left ventricular afterload should be avoided.
Secondary Prevention
Effective measures for the secondary prevention of hypertrophic cardiomyopathy include screening. Once HCM has been identified in a family, immediate testing of all family members will help to identify those at risk. Both imaging and genetic testing might be helpful. Athletes and military commanders (this in danger group barely discussed in the literature) are particularly in danger and it is recommended to undergo screening for HCM.
Management During Pregnancy
Women with hypertrophic cardiomyopathy should be managed by a skilled cardiovascular specialist and a high-risk obstetrician during pregnancy. Among HCM patients who chronically have mild symptoms, pregnancy is generally well tolerated [1][2]. Although pregnancy causes vasodilation which should exacerbate the outflow gradient, pregnancy also causes fluid retention and an increase in plasma volume which increases preload and offsets the reduction in afterload. In a series of 100 HCM patients, only one of 28 asymptomatic patients developed NYHA Class III or IV heart failure.
Epidural Anesthesia Should Be Avoided due to the potential for venous pooling. Bleeding should be minimized. Blood should be crossed and typed in case a transfusion is needed for bleeding, which can exacerbate outflow obstruction. Although both beta blockers and verapamil may improve symptoms in the mother, the dosing should be limited to minimize the risk of fetal bradycardia, growth retardation and hypoglycemia. There is more experience with the use beta blockers during pregnancy. Home delivery without IV access is not preferred. Vaginal delivery is usually successful.
References
- ↑ Oakley GD, McGarry K, Limb DG, Oakley CM (1979). “Management of pregnancy in patients with hypertrophic cardiomyopathy”. British Medical Journal. 1 (6180): 1749–50. PMC 1599373. PMID 572730. Unknown parameter
|month=ignored (help) - ↑ Autore C, Conte MR, Piccininno M, Bernabò P, Bonfiglio G, Bruzzi P, Spirito P (2002). “Risk associated with pregnancy in hypertrophic cardiomyopathy”. Journal of the American College of Cardiology. 40 (10): 1864–9. PMID 12446072. Unknown parameter
|month=ignored (help)
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Soroush Seifirad, M.D.[2]
Overview
The first case of hypertrophic cardiomyopathy (HCM) was described by in 1869 Henri Liouville in the Gazette Medecine Paris. In 1907 Dr. A. Schmincke, a German pathologist, described two hearts with left ventricular hypertrophy; both came from women in their mid-fifties. Levy and von Glahn in 1944, from Colombia University in New York, published a series of cases which resembles HCM. In 1949, William Evans, a London cardiologist, described familial occurrence of cardiac hypertrophy in a series of patients which were similar to those described in the paper by Levy and von Glahn. In 1961 Paré et al. reported thirty members of five generations of a French Canadian family in Quebec in whom the condition was inherited in an autosomal dominant manner. In 1958 Teare, an English pathologist, described eight cases of asymmetric cardiac muscle hypertrophy, he thought that they might be benign cardiac tumors. Seven of these caused sudden death in young adults. Teare named the condition “Asymetrical Hypertrophy of the Heart.” In 1959 Sir Russell Brock described a young man with angina and a subaortic stenosis and a subaortic intraventricular pressure gradient. Morrow and Braunwald published their first report in the same year, followed by several other reports. The sudden cardiac deaths of 387 young American athletes (under age 35) were analyzed in a 2003 medical review, and HCM was the leading cause of sudden cardiac death in athletes. In 1961, Morrow described a surgical procedure to relieve the obstruction, which is still the most widely used method of surgical treatment. In 1962, with respect to the observed intensification of obstruction in HCM with the beta-adrenergic agonists, Braunwald suggested the use of newly developed beta-blockers. In 1964, Braunwald reported beta-blockers beneficial hemodynamic effects. 1967, the clinical benefits of treatment with beta blockers in patients with HCM has been proved to the scientific society.
Historical Perspective
Discovery
- The first case of hypertrophic cardiomyopathy (HCM) was described by in 1869 Henri Liouville in the Gazette Medecine Paris. He described a 75-year-old woman who developed worsening dyspnea over several days. On physical examination, she had a systolic heart murmur. She died shortly after the presentation.[1] Liouville H. Rétrécissement cardiaque sous aortique. Gazette Medecine Paris. 1869;24:161–163.
- In 1907 Dr. A. Schmincke, a German pathologist, described two hearts with left ventricular hypertrophy; both came from women in their mid fifties. [1]Schmincke A. Ueber linkseitige muskulose conustenosen. Deutsche Med Wochenschr. 1907;33:2082–2085.
- Levy and von Glahn in 1944, from Colombia University in New York, published a series of cases which resembles HCM.[1] Levy RL, von Glahn WC. Cardiac hypertrophy of unknown cause. A study of the clinical and pathologic features in ten adults. Am Heart J. 1944;28:714–741.
- In 1949, William Evans, a London cardiologist, described familial occurrence of cardiac hypertrophy in a series of patients which were similar to those described in the paper by Levy and von Glahn. [2]
- In 1961 Paré et al. reported thirty members of five generations of a French Canadian family in Quebec in whom the condition was inherited in an autosomal dominant manner.[3]
- In 1958 Teare, an English pathologist, described eight cases of asymmetric cardiac muscle hypertrophy, he thought that they might be benign cardiac tumors. Seven of these caused sudden death in young adults. Teare named the condition “Asymetrical Hypertrophy of the Heart.”[4]
- In 1959 Sir Russell Brock described a young man with angina and a subaortic stenosis and a subaortic intraventricular pressure gradient. He mentioned: “That this is not an isolated case is made clear by the experience of Dr. Glenn Morrow who tells me he has operated on two similar cases in two young men in their early twenties; both survived. He has kindly allowed me to mention these prior to his own report of them (Morrow and Braunwald, Circulation, in press, 1959).” [5]
- Morrow and Braunwald published their first report in the same year, followed by several other reports. [6]
Landmark Events in the Development of Treatment Strategies
- In 1961, Morrow described a surgical procedure (myectomy) to relieve the obstruction,[7] which is still the most widely used method of surgical treatment.
- In 1962, with respect to the observed intensification of obstruction in HCM with the beta-adrenergic agonists,[8] Braunwald suggested the use of newly developed beta-blockers.
- In 1964, Braunwald reported beta-blockers beneficial hemodynamic effects.[9]
- In 1967, the clinical benefits of treatment with beta-blockers in patients with HCM has been proved to the scientific society. [10][11][1]
Impact on Cultural History
Famous Cases
The following are a few famous cases of hypertrophic cardiomyopathy:
- Sir David Paradine Frost: A post-mortem following the death of popular TV presenter David Frost in 2013 showed he suffered from HCM, though it didn’t contribute to his death and his family wasn’t informed. The sudden cardiac death of his 31-year-old son in 2015 led the family to collaborate with the British Heart Foundation to raise funds for better screening.
- The sudden cardiac deaths of 387 young American athletes (under age 35) were analyzed in a 2003 medical review, and HCM was the leading cause of sudden cardiac death in athletes.[12] Hence, there is a lengthy list of athletes that passed away because of HCM, among them are:
- Reggie Lewis, Baltimore Basketball Player
- Gaines Adams, American professional football player
- Heath Benedict, Dutch-American professional football player
- James Victor Cain, American professional football player
- Mitchell Cole, English footballer (Soccer)
References
- ↑ 1.0 1.1 1.2 1.3 Braunwald E (2012). “Hypertrophic cardiomyopathy: The first century 1869-1969”. Glob Cardiol Sci Pract. 2012 (1): 5. doi:10.5339/gcsp.2012.5. PMC 4239819. PMID 25610836.
- ↑ EVANS W (1949). “Familial cardiomegaly”. Br Heart J. 11 (1): 68–82. doi:10.1136/hrt.11.1.68. PMC 503618. PMID 18113470.
- ↑ PARE JA, FRASER RG, PIROZYNSKI WJ, SHANKS JA, STUBINGTON D (1961). “Hereditary cardiovascular dysplasia. A form of familial cardiomyopathy”. Am J Med. 31: 37–62. doi:10.1016/0002-9343(61)90222-4. PMID 13732753.
- ↑ TEARE D (1958). “Asymmetrical hypertrophy of the heart in young adults”. Br Heart J. 20 (1): 1–8. doi:10.1136/hrt.20.1.1. PMC 492780. PMID 13499764.
- ↑ BROCK R (1959). “Functional obstruction of the left ventricle (acquired aortic subvalvar stenosis)”. Guys Hosp Rep. 108: 126–43. PMID 13804574.
- ↑ MORROW AG, BRAUNWALD E (1959). “Functional aortic stenosis; a malformation characterized by resistance to left ventricular outflow without anatomic obstruction”. Circulation. 20 (2): 181–9. doi:10.1161/01.cir.20.2.181. PMID 13671704.
- ↑ MORROW AG, BROCKENBROUGH EC (1961). “Surgical treatment of idiopathic hypertrophic subaortic stenosis: technic and hemodynamic results of subaortic ventriculomyotomy”. Ann Surg. 154: 181–9. doi:10.1097/00000658-196108000-00003. PMC 1465878. PMID 13772904.
- ↑ BRAUNWALD E, EBERT PA (1962). “Hemogynamic alterations in idiopathic hypertrophic subaortic stenosis induced by sympathomimetic drugs”. Am J Cardiol. 10: 489–95. doi:10.1016/0002-9149(62)90373-9. PMID 14015086.
- ↑ HARRISON DC, BRAUNWALD E, GLICK G, MASON DT, CHIDSEY CA, ROSS J (1964). “EFFECTS OF BETA ADRENERGIC BLOCKADE ON THE CIRCULATION WITH PARTICULAR REFERENCE TO OBSERVATIONS IN PATIENTS WITH HYPERTROPHIC SUBAORTIC STENOSIS”. Circulation. 29: 84–98. doi:10.1161/01.cir.29.1.84. PMID 14105035.
- ↑ Cohen LS, Braunwald E (1967). “Amelioration of angina pectoris in idiopathic hypertrophic subaortic stenosis with beta-adrenergic blockade”. Circulation. 35 (5): 847–51. doi:10.1161/01.cir.35.5.847. PMID 6067064.
- ↑ Braunwald E (2009). “Hypertrophic cardiomyopathy: the early years”. J Cardiovasc Transl Res. 2 (4): 341–8. doi:10.1007/s12265-009-9128-3. PMID 20559993.
- ↑ Maron BJ (2003). “Sudden death in young athletes”. N Engl J Med. 349 (11): 1064–75. doi:10.1056/NEJMra022783. PMID 12968091.
Classification
Editors-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Soroush Seifirad, M.D.[2]
Overview
There are two variants of hypertrophic cardiomyopathy: an obstructive variant, and a non-obstructive variant. About 25% of individuals with hypertrophic cardiomyopathy (HCM) demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In other individuals, obstruction only occurs under certain conditions. This is known as dynamic outflow obstruction because the degree of obstruction is variable and is dependent on the amount of blood in the ventricle immediately before ventricle systole (contraction). If left ventricular outflow obstruction is present, then this syndrome has been known as wide variety of terms including: hypertrophic cardiomyopathy or HCM, asymmetric septal hypertrophy or ASH, hypertrophic obstructive cardiomyopathy, HOCM, idiopathic hypertrophic subaortic stenosis or IHSS.
A non-obstructive variant of HCM is known as apical hypertrophic cardiomyopathy , which is also known as nonobstructive hypertrophic cardiomyopathy and Japanese variant hypertrophic cardiomyopathy or the Yamaguchi variant (since the first cases described were all in individuals of Japanese descent), also known as apical hypertrophic cardiomyopathy (ApHCM) or Yamaguchi syndrome.
Obstructive Variant
About 25% of individuals with hypertrophic cardiomyopathy (HCM) demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In other individuals, obstruction only occurs under certain conditions. This is known as dynamic outflow obstruction because the degree of obstruction is variable and is dependent on the amount of blood in the ventricle immediately before ventricle systole (contraction). If left ventricular outflow obstruction is present, then this syndrome has been known as wide variety of terms including: hypertrophic cardiomyopathy or HCM, asymmetric septal hypertrophy or ASH, hypertrophic obstructive cardiomyopathy, HOCM, idiopathic hypertrophic subaortic stenosis or IHSS, familial isolated hypertrophic obstructive cardiomyopathy, familial isolated hypertrophic subaortic stenosis, familial or idiopathic hypertrophic subaortic stenosis, familial or idiopathic hypertrophic obstructive cardiomyopathy, primitive hypertrophic obstructive cardiomyopathy, primitive hypertrophic subaortic stenosis.



Non-Obstructive Variant
A non-obstructive variant of HCM is known as apical hypertrophic cardiomyopathy [1], which is also known as nonobstructive hypertrophic cardiomyopathy and Japanese variant hypertrophic cardiomyopathy or the Yamaguchi variant (since the first cases described were all in individuals of Japanese descent), also known as apical hypertrophic cardiomyopathy (ApHCM) or Yamaguchi syndrome.
- Ace of spade observation in echocardiography is pathognomonic of apical hypertrophy.
- Nevertheless, hypertrophic non-obstructive cardiomyopathy can either be familial or sporadic. It has been shown that a number of chronic medical conditions might be contributed to HNCM development such as thyroid disease, diabetes, and obesity.
- Mutations in several genes (MYH7, MYBPC3, TNNT2, and TNNI3 genes) cause the inherited form of hypertrophic cardiomyopathy.
- This is an autosomal dominant familial disorder.
- HNCM can potentially evolve into:
- Hypertrophic obstructive cardiomyopathy
- Heart valve regurgitation
- Aberrant heartbeats (arrhythmia)
- Sudden cardiac arrest
- Dilated cardiomyopathy
References
- ↑ Rivera-Diaz J, Moosvi AR. Apical hypertrophic cardiomyopathy. South Med J. 1996 Jul; 89(7):711-3. (Medline abstract; Full text)
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Cafer Zorkun, Soroush Seifirad, M.D.[2]
Overview
The progression to hypertrophic cardiomyopathy usually involves the mutations in contractile sarcomeric proteins of myocardium, which describe the presence of left ventricular hypertrophy (LVH) in the absence of an increased external load (unexplained LVH). Additionally, HCM hypertrophy is generally asymmetric.
HCM is the most common genetically transmitted cardiovascular disease. Hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins. Penetrance of HCM is incomplete, variable and time or age-related. The disease may be sporadic but affected family members are discovered in 13% of cases. More than 200 mutations involving at least 10 chromosomes encoding structural proteins of the myocyte have been discovered. These mutations have varying degrees of penetrance and even the same mutation may have variable expression, implying the superimposed effects of other genes or environmental influences. Children of a patient with HCM have a 50% chance of inheriting the trait.
Depending on the degree of obstruction of the outflow of blood from the left ventricle of the heart, HCM can be defined as obstructive or non-obstructive. About 25% of individuals with HCM demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In other individuals, obstruction only occurs under certain conditions. This is known as dynamic outflow obstruction because the degree of obstruction is variable and is dependent on the amount of blood in the ventricle immediately before ventricle systole (contraction).
Although there may be structural or functional obstruction of the left ventricular outflow tract, symptoms may arise more often from diastolic dysfunction.There is extensive periarteriolar fibrosis that results in microvascular dysfunction and impairment in coronary flow reserve in patients with hypertrophic obstructive cardiomyopathy. Individuals with HCM have some degree of left ventricular hypertrophy. In approximately 2/3rds of cases this is asymmetric hypertrophy, involving the interventricular septum, and is known as asymmetric septal hypertrophy (ASH). This is in contrast to the symmetric and concentric hypertrophy seen in aortic stenosis or hypertension. On histopathologic examination, hypertrophic cardiomyopathy is characterized by both myocardial disarrays and by periarteriolar fibrosis. Myocardial disarray can be associated with aberrant impulse conduction and arrhythmias, and periarteriolar fibrosis can be associated with myocardial ischemia.
Pathophysiology
Physiology
The normal physiology of myocardium can be understood as follows:
- The myocardium is composed of specialized cardiac muscle cells with an ability not possessed by muscle tissue elsewhere in the body. Cardiac muscle, like other muscles, can contract, but it can also carry an action potential (i.e. conduct electricity), like the neurones that constitute nerves.
Pathogenesis
- The progression to Hypertrophic cardiomyopathy usually involves the mutations in contractile sarcomeric proteins of myocardium, which describe the presence of left ventricular hypertrophy (LVH) in the absence of an increased external load (unexplained LVH).
- Additionally, HCM hypertrophy is generally asymmetric.
Genetics
Hypertrophic cardiomyopathy is transmitted in an autosomal dominant pattern.
Genes involved in the pathogenesis of hypertrophic cardiomyopathy include:
The development of hypertrophic cardiomyopathy is the result of multiple genetic mutations such as:
- Beta-myosin heavy chain
- Myosin binding protein C
- Cardiac troponin T
HCM is the most common genetically transmitted cardiovascular disease. Hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins. [1][2][3][4][5][6][7][8][9][10][11][12][13][14][15] Penetrance of HCM is incomplete, variable and time or age-related. The disease may be sporadic but affected family members are discovered in 13% of cases. More than 200 mutations involving at least 10 chromosomes encoding structural proteins of the myocyte have been discovered. These mutations have varying degrees of penetrance and even the same mutation may have variable expression, implying superimposed effects of other genes or environmental influences. Children of a patient with HCM have a 50% chance of inheriting the trait.
Mutations
Common Mutations
Mutations in three regions affect more than half the patients with HCM:
- Beta-myosin heavy chain
- Myosin binding protein C
- Cardiac troponin T
Complete List of Mutations
Hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins including beta-cardiac myosin heavy chain (the first gene identified), cardiac actin, cardiac troponin T, alpha-tropomyosin, cardiac troponin I, cardiac myosin-binding protein C, and the myosin light chains. Specific gene mutations that have been identified include the following:
| Gene | Locus | Type |
|---|---|---|
| MYH7 | 14q12 | CMH1 |
| TNNT2 | 1q32 | CMH2 |
| TPM1 | 15q22.1 | CMH3 (115196) |
| MYBPC3 | 11p11.2 | CMH4 (115197) |
| ? | ? | CMH5 |
| PRKAG2 | 7q36 | CMH6 (600858) |
| TNNI3 | 19q13.4 | CMH7 |
| MYL3 | 3p | CMH8 (608751) |
| TTN | 2q24.3 | CMH9 |
| MYL2 | 12q23-q24 | CMH10 |
| ACTC1 | 15q14 | CMH11 (612098) |
| CSRP3 | 11p15.1 | CMH12 (612124) |
While the above table represents the most common genetic mutations, there are also about 200 intergenic (within a gene) mutations. These include missense and single amino acid residue substitutions. There are different genetic mutations in different families. The environment may also play a role because affected individuals in the same family may have a different phenotypic expression (i.e different degrees of left ventricular hypertrophy). The goal of modifier genes in regulating phenotypic expression is not clear.
While genes, gene modifiers, and environment may play a role in the phenotypic expression of left ventricular hypertrophy, genes may also play a role in the risk of arrhythmias. While most literature so far focuses on European, American, and Japanese populations, HCM appears in all races. The incidence of HCM is about 0.2% to 0.5% of the general population.
Specific Chromosomal Abnormalities
β Myosin Heavy Chain-Chromosome 14 q11.2-3
In individuals without a family history of HCM, the most common cause of the disease is a de novo mutation of the gene that produces the β-myosin heavy chain. This chromosomal abnormality accounts for approximately 35%-45% of HCM cases. Significant LVH (left ventricular hypertrophy) is usually present. The Arg403Gln mutation is associated with an extremely poor prognosis with an average age of death at 33 years, while the Val606Met mutation is associated with a better prognosis.
Cardiac Troponin T-Chromosome 11
Accounts for approximately 15% of cases. Substantially less hypertrophy is noted but histology demonstrates the characteristic myocyte disarray of HCM. Most mutations of this gene are associated with markedly reduced survival.
Cardiac Myosin Binding Protein-C-Chromosome 11
This chromosomal abnormality accounts for 15% to 35% of patients, but given the reduced penetrance associated with this abnormality, the true incidence may actually be greater. Patients generally present later in life and in general, have a better prognosis than beta myosin heavy chain or cardiac troponin T mutations. Up to 60% of patients at age 50 years have no evidence of LVH. LVH may appear later in life in these patients. Because of this, a normal EKG and a normal echocardiography at age 18 does not exclude the presence of HCM.
Arg663 His mutation
The beta-myosin heavy chain Arg663 His mutation is associated with a higher risk of atrial fibrillation. [16]
PRKAG2 Mutation
There is no myocyte disarray, but the conduction block is present. This variant is more akin to a storage disease.[17]
Mutations that Alter the Phenotypic Expression of the Disease
An insertion/deletion polymorphism in the gene encoding for angiotensin converting enzyme (ACE) alters the clinical phenotype of the disease. The D/D (deletion/deletion) genotype of ACE is associated with more marked hypertrophy of the left ventricle and may be associated with higher risk of adverse outcomes. [18] [19]
Genetic Testing
Whenever a mutation is identified through genetic testing, family-specific genetic testing can be used to identify relatives at-risk for the disease (HCM Genetic Testing Overview). In individuals without a family history of HCM, the most common cause of the disease is a de novo mutation of the gene that produces the β-myosin heavy chain.
Outflow Obstruction
Depending on the degree of obstruction of the outflow of blood from the left ventricle of the heart, HCM can be defined as obstructive or non-obstructive. About 25% of individuals with HCM demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In other individuals obstruction only occurs under certain conditions. This is known as dynamic outflow obstruction, because the degree of obstruction is variable and is dependent on the amount of blood in the ventricle immediately before ventricle systole (contraction).
Location Of The Left Ventricular Outflow Obstruction
The left ventricular obstruction can be either
- Mid-cavitary: the middle of the ventricle or
- Sub-aortic: just below the aortic valve
Classification of the Valve Gradient in Hypertrophic Cardiomyopathy
The valve gradient in HCM can be classified into three categories:
- A gradient greater than 30 mm Mercury under basal conditions
- A gradient that is greater than 30 mm Mercury with provocation
- A gradient that is less than 30 mm Mercury at rest and with provocation
Maneuvers that Increase the Outflow Gradient
- Amyl nitrite inhalation
- Valsalva maneuver
- Premature ventricular contractions (PVCs)
- Isoproterenol infusion
- Dobutamine infusion; but this is not recommended as a diagnostic tool[20][21]
- Treadmill or exercise stress testing
Causes of Left Ventricular Outflow Obstruction: Systolic Anterior Motion of the Mitral Valve (SAM)
If dynamic outflow obstruction is present in a patient with HCM, it is usually due to systolic anterior motion (SAM) of the anterior leaflet of the mitral valve. The systolic anterior motion of the mitral valve (SAM) may be due to a subaortic bulge of the septum along with narrowing the left ventricular outflow tract, which taken together cause high-velocity flow. This, in turn, is associated with the Venturi effect which is a local low-pressure zone in the left ventricular outflow tract. This low-pressure zone was thought to suck the mitral valve anteriorly into the septum. More recently, however, SAM onset has been observed to be instead a low-velocity phenomenon. [22] [23] The role of Venturi forces in the left ventricular outflow tract may be less important than previously thought. While the Venturi effect was thought to cause the abnormality in prior studies, more recent echocardiographic studies indicates that drag, which is more of a pushing force rather than a sucking force like the Venturi effect, maybe the dominant hydrodynamic force acting on the mitral leaflets.[22][23][24][25][26][27]
The videos below show examples of systolic anterior motion of the mitral valve:
{{#ev:youtube|Y7JUVTXHBs0}}
{{#ev:youtube|6TWb-wIL0H0}}
Impact of Systolic Anterior Motion of the Mitral Valve: The Spike and Dome Pattern to the Carotid Pulse
Because the mitral valve leaflet doesn’t get pulled into the left ventricular outflow tract (LVOT) until after the aortic valve opens, the initial upstroke of the arterial pulse pressure will be normal. When the mitral valve leaflet gets pushed into the LVOT, the arterial pulse will momentarily collapse and will later be followed by a second rise in the pulse pressure, as the left ventricular pressure overcomes the increased obstruction caused by the SAM of the mitral valve. This can be seen on the physical examination as a double-tap upon palpation of the apical impulse and as a double pulsation upon palpation of the carotid pulse, known as pulsus bisferiens or a “spike and dome pattern” to the carotid pulse.
Accompanying Mitral Regurgitation
As a result of the drag effect or the Venturi effect, there may be mild to moderate mitral regurgitation in association with hypertrophic cardiomyopathy. Most often the mitral regurgitation jet is directed posteriorly. If the jet is not directed posteriorly then other diagnoses should be considered which include myxomatous degeneration or other anomalies of the mitral valve.
Pathophysiologic Consequences of Outflow Obstruction
Chronic outflow obstruction and result in the following abnormalities:[28][29]
- Increased left ventricular wall stress
- Myocardial ischemia
- Myocardial necrosis
- Replacement fibrosis
Prognostic Significance of Outflow Obstruction
The presence of outflow obstruction is associated with a twofold increased risk of death and a 4.4 fold increase in the risk of progression to New York Heart Association class III or IV heart failure. [30][31] Above a gradient of 30 mm Hg, there was no further increase in the risk of sudden cardiac death or progression of congestive heart failure symptoms.[32]
Ischemia
There is extensive periarteriolar fibrosis that results in microvascular dysfunction and impairment in coronary flow reserve in patients with hypertrophic obstructive cardiomyopathy.
Histopathology
Compared to normal arterioles on the left, the arterioles from a patient with hyertension (middle) show moderate periarteriolar thickening and fibrosis. Shown on the right is a patient with HCM in which there is even more signficant periarteriolar thickening and fibrosis. This thickening of the wall of the intramyocardial arterioles leads to an increased wall/lumen ratio, subendocardial ischemia and impaired coronary flow reserve.[33][34] Patients who subsequently died in one series had abnormal coronary flow reserve on PET scanning at baseline indicating that ischemia may play a role, at least in part, in subsequent mortality.
-
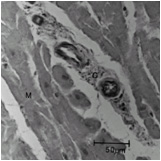 Normal arteriole
Normal arteriole -
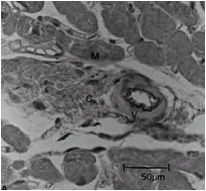 Hypertensive arteriole with wall thickening and myocyte hypertrophy
Hypertensive arteriole with wall thickening and myocyte hypertrophy -
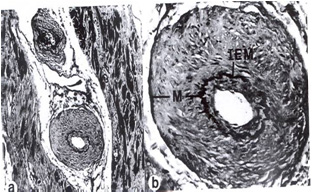 Arteriole in HCM patient with periarteriole fibrosis and thicknening
Arteriole in HCM patient with periarteriole fibrosis and thicknening



Arrhythmogenesis
Patients with hypertrophic cardiomyopathy are at risk of arrhythmias and sudden death. Abnormal filling of the left atrium may result in the left atrial dilation which may predispose the patient to atrial fibrillation. The presence of myocardial disarray and myocardial ischemia (due to microvascular dysfunction and episodes of reduced cardiac output) may predispose the patient to ventricular tachycardia, ventricular fibrillation, and sudden cardiac death.
Atrial Arrhythmias
Impaired filling of the left ventricle can lead to left atrial stretch and left atrial dilation. This, in turn, can predispose the patient to the development of atrial fibrillation. The onset of atrial fibrillation can be quite dangerous in these patients as the loss of left atrial kick and the more rapid heart rate can both diminish left ventricular filling which can lead to severe hemodynamic compromise. This hemodynamic compromise can, in turn, be associated with sudden cardiac death.
Ventricular Arrhythmias
Ventricular arrhythmias and degeneration into sudden cardiac death may be due to the following:
- Primary arrhythmias
- The presence of myocardial disarray
- The presence of scar or fibrosis
- Hemodynamic instability with diminished stroke volume
- The presence of ischemia
It must be emphasized that atrial arrhythmias (which are commonly detected on ambulatory monitoring) can lead to ischemia and hemodynamic compromise which may, in turn, lead to sudden cardiac death in these patients as well.
Autonomic Imbalance
Assessment of autonomic function in patients with HCM often reveals abnormal responses of heart rate and blood pressure to exercise in two-thirds, which was associated with a more malignant clinical course, suggesting that autonomic imbalance may also be important in the genesis of sudden cardiac death in these patients.
Anatomic abnormalities
Individuals with HCM have some degree of left ventricular hypertrophy. In approximately 2/3rds of cases this is asymmetric hypertrophy, involving the interventricular septum, and is known as asymmetric septal hypertrophy (ASH). This is in contrast to the symmetric and concentric hypertrophy seen in aortic stenosis or hypertension.
Left Ventricle
The degree of ventricular hypertrophy is variable ranging from diffuse involvement of both ventricles to isolated involvement of a portion of one segment of the LV.
Data from two large registries indicate that;
- 55% of cases involve the septum and anterolateral free wall,
- 20% involve the entire septum alone,
- 10% is limited to the nasal septum and 15% are limited to the apical or distal LV (Yamaguchi variant).
Some genetic variants may manifest very little overt LVH but are still associated with an increased risk of sudden cardiac death (SCD).
Outflow Tract
The left ventricular outflow tract is often small.
Mitral Valve
The mitral valve maybe elongated and enlarged.
Associated Conditions
Conditions associated with Hypertrophic cardiomyopathy include:
- [Condition 1]
- [Condition 2]
- [Condition 3]
Histopathologic Abnormalities
On histopathologic examination, hypertrophic cardiomyopathy is characterized by both myocardial disarrays and by periarteriolar fibrosis. Myocardial disarray can be associated with aberrant impulse conduction and arrhythmias, and periarteriolar fibrosis can be associated with myocardial ischemia.
Myocardial Disarray
In HCM, the normal alignment of muscle cells is disrupted (there is a swirling pattern to the arrangement of the muscle cells), a phenomenon known as myocardial disarray. HCM is believed to be due to a mutation in one of many genes that results in a mutated myosin heavy chain, one of the components of the myocyte (the muscle cell of the heart). Histopathologically, the cardiac sarcomere is abnormal resulting in hypertrophy of the left ventricle in the absence of other disorders that could produce the condition such as hypertension, amyloid or aortic stenosis. The presence of myocardial disarray may be associated with abnormalities of electrical conduction in the heart (including electrical reentry loops) which thereby contributes to an increased risk of sudden cardiac death.
-
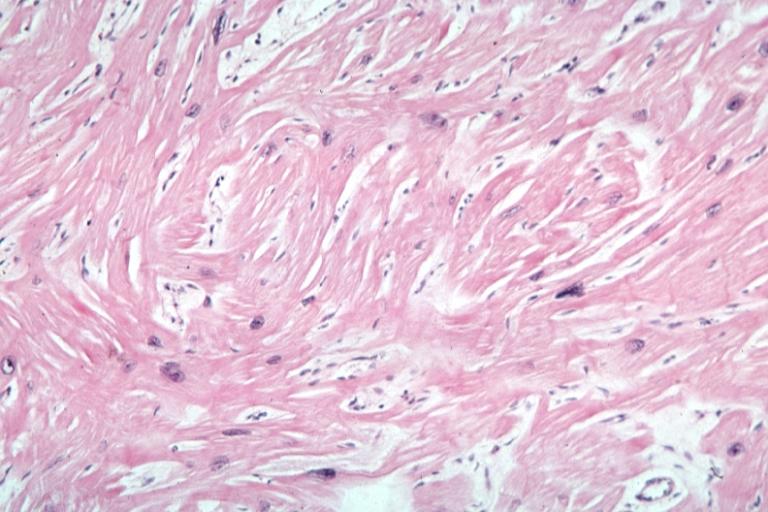 Myocardial disarray with swirling pattern of myocytes
Myocardial disarray with swirling pattern of myocytes -
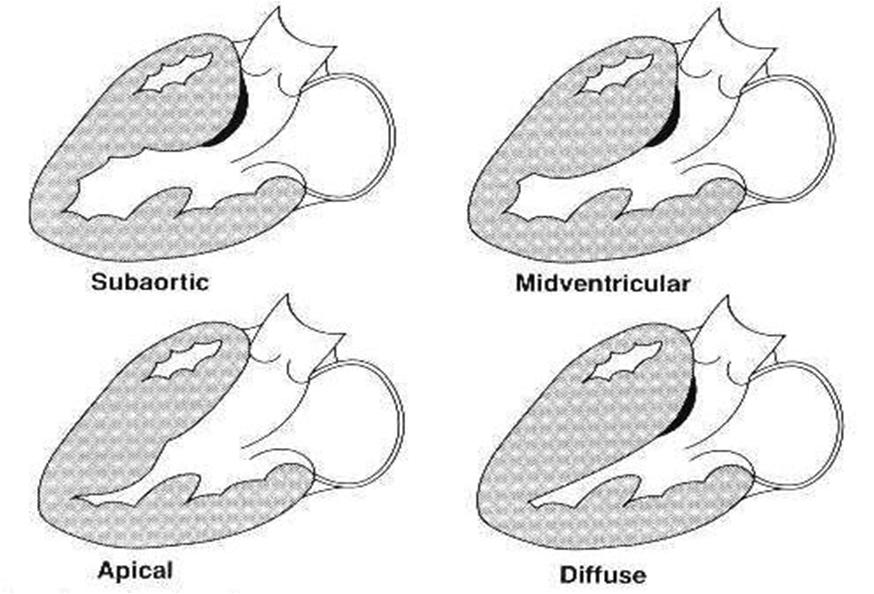 Variants of hypertrophic cardiomyopathy
Variants of hypertrophic cardiomyopathy -
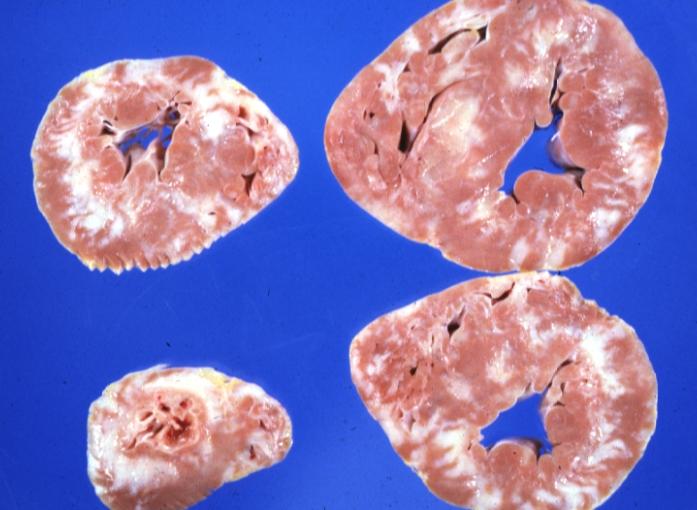 White areas of fibrosis or scar in a patient with HCM which may contribute in part to arrhythmias
White areas of fibrosis or scar in a patient with HCM which may contribute in part to arrhythmias



Periarteriolar Fibrosis
Compared to normal arterioles on the left, the arterioles from a patient with hyertension (middle) show moderate periarteriolar thickening and fibrosis. Shown on the right is a patient with HCM in which there is even more signficant periarteriolar thickening and fibrosis. This thickening of the wall of the intramyocardial arterioles leads to an increased wall/lumen ratio, subendocardial ischemia and impaired coronary flow reserve.[33][34] Patients who subsequently died in one series had abnormal coronary flow reserve on PET scanning at baseline indicating that ischemia may play a role, at least in part, in subsequent mortality.
-
Normal arteriole
-
Hypertensive arteriole with wall thickening and myocyte hypertrophy
-
Arteriole in HCM patient with periarteriole fibrosis and thicknening
Gross Pathology
On gross pathology, asymmetric interventricular wall thickening is characteristic findings of hypertrophic cardiomyopathy. Subaortic stenosis could be evident in many cases. In the Yamaguchi subtype, there is apical hypertrophy.
Microscopic Pathology
On microscopic histopathological analysis, myocardial disarray, periarteriolar fibrosis, and hypertrophy are characteristic findings of hypertrophic cardiomyopathy.
Histopathologically, small vessels have hypertrophy of the tunica media. Combined with increased wall tension, decreased vasodilator reserve, and inadequate capillary density, there is a mismatch between blood supply and demand. Over time, it is thought that there is repeated ischemia followed by fibrosis and eventually, dilation and systolic dysfunction (“burned out hypertrophy”).
-
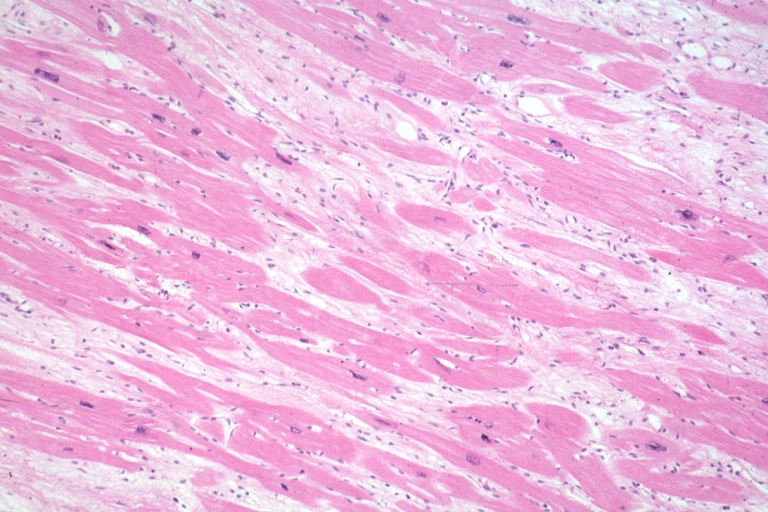 Micro med mag H&E mid-mural myocardium with hypertrophy and interstitial fibrosis atrophy is present marked increase in interstitial fibroblastic cells
Micro med mag H&E mid-mural myocardium with hypertrophy and interstitial fibrosis atrophy is present marked increase in interstitial fibroblastic cells -
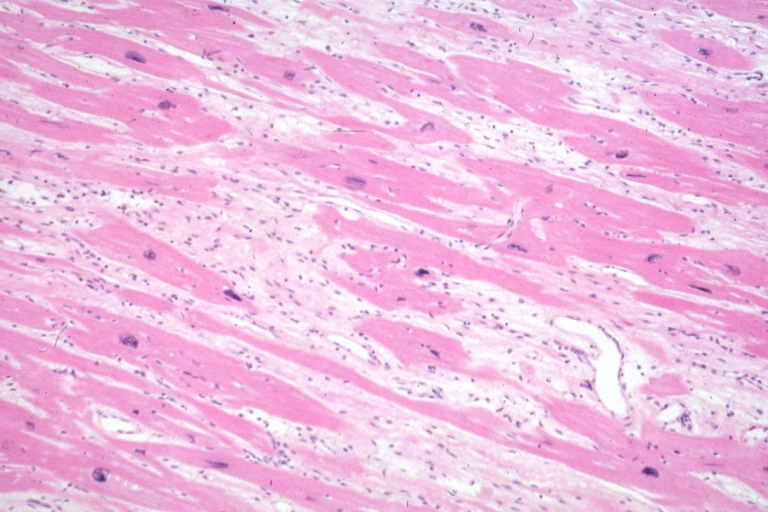 Micro high mag H&E myofiber hypertrophy and interstitial fibrosis with marked increase in interstitial fibroblastic cells
Micro high mag H&E myofiber hypertrophy and interstitial fibrosis with marked increase in interstitial fibroblastic cells


-
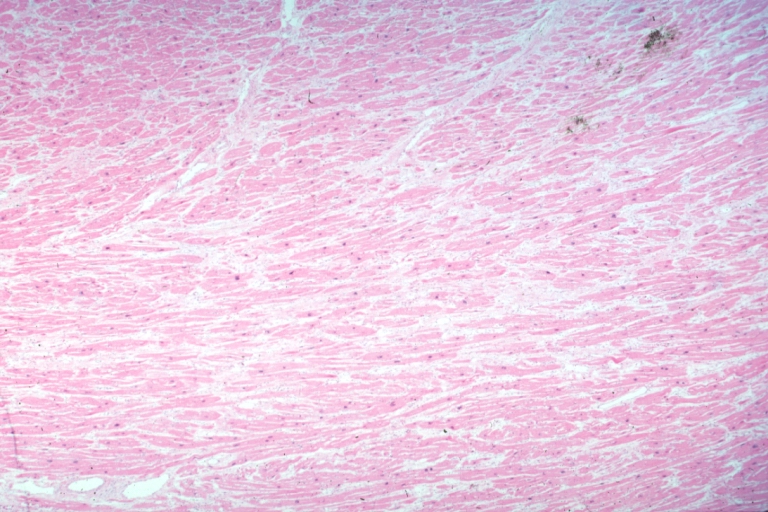 Micro med mag H&E myofiber hypertrophy some atrophy interstitial fibrosis with many fibroblastic cells
Micro med mag H&E myofiber hypertrophy some atrophy interstitial fibrosis with many fibroblastic cells -
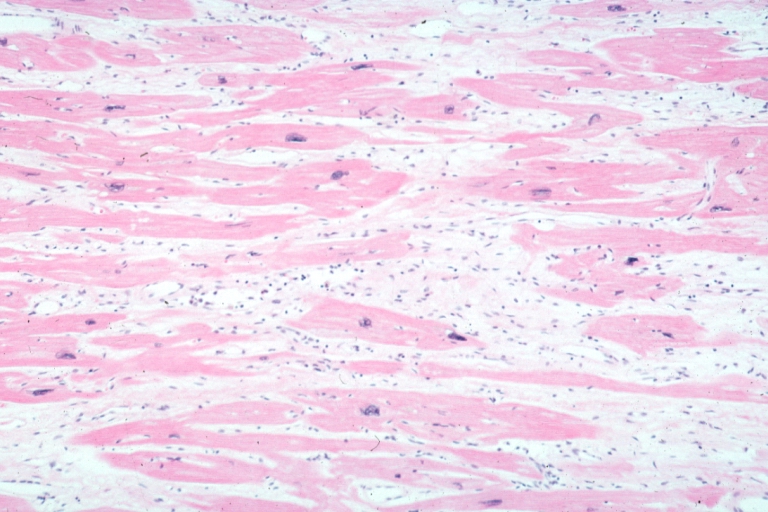 Micro high mag H&E hypertrophied fibers with some evidence of atrophy and marked interstitial fibrosis with many fibroblastic type cells
Micro high mag H&E hypertrophied fibers with some evidence of atrophy and marked interstitial fibrosis with many fibroblastic type cells


-
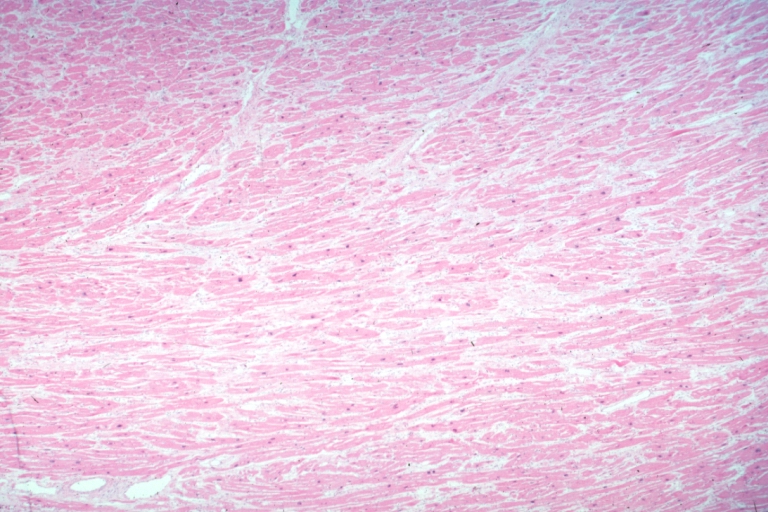 Micro low mag H&E shows myofiber hypertrophy and interstitial fibrosis
Micro low mag H&E shows myofiber hypertrophy and interstitial fibrosis -
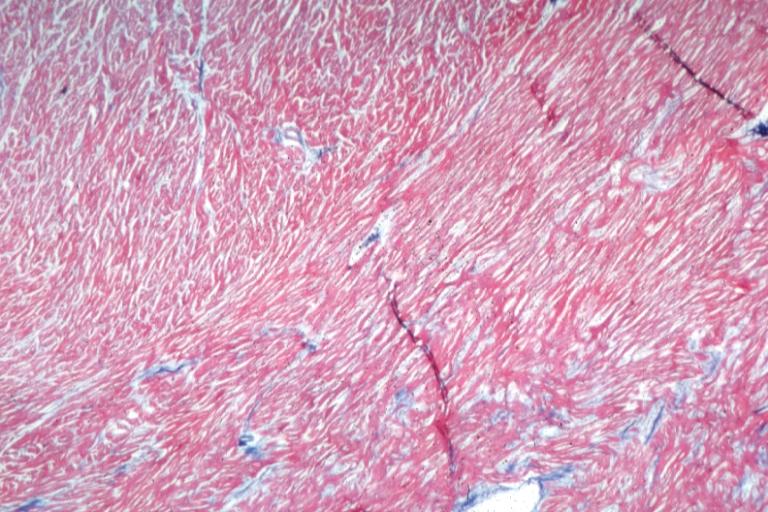 Cardiomyopathy: Micro H&E low mag interventricular septum at junction of normal myofiber orientation with asymmetrical hypertrophy (an excellent example)
Cardiomyopathy: Micro H&E low mag interventricular septum at junction of normal myofiber orientation with asymmetrical hypertrophy (an excellent example)


-
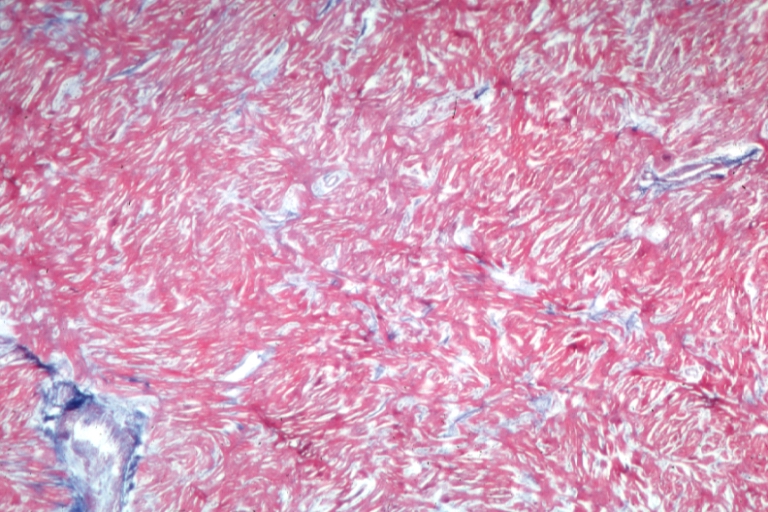 Cardiomyopathy: Micro H&E low mag marked myofiber disarray asymmetrical hypertrophy
Cardiomyopathy: Micro H&E low mag marked myofiber disarray asymmetrical hypertrophy -
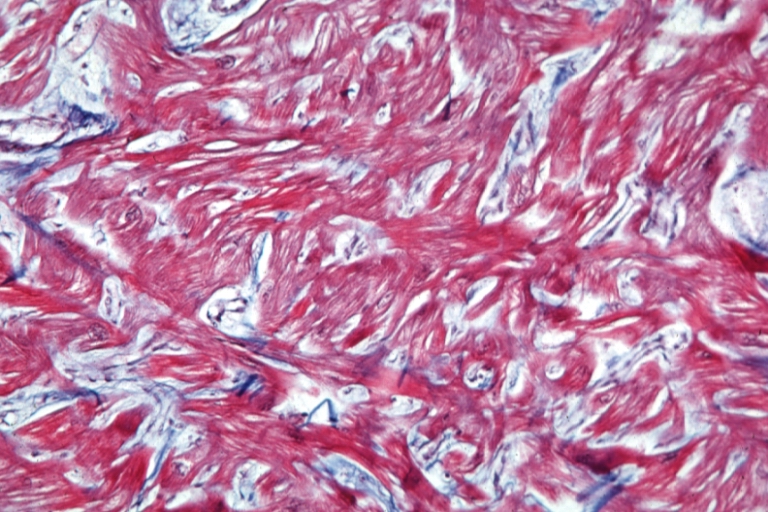 Cardiomyopathy: Micro trichrome high mag marked myofiber disarray
Cardiomyopathy: Micro trichrome high mag marked myofiber disarray


-
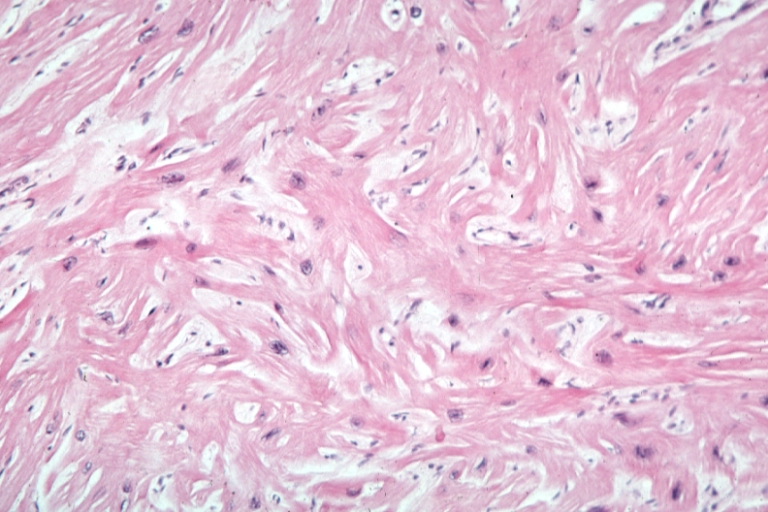 Cardiomyopathy: Micro H&E med mag excellent example myofiber disarray
Cardiomyopathy: Micro H&E med mag excellent example myofiber disarray -
Cardiomyopathy: Micro H&E high mag excellent example myofiber disarray

References
- ↑ Maron BJ, Moller JH, Seidman CE et al. Impact of laboratory molecular diagnosis on contemporary diagnostic criteria for genetically transmitted cardiovascular diseases. Hypertrophic cardiomyopathy, long-QT syndrome, and Marfan syndrome. [A statement for healthcare professionals from the Councils on Clinical Cardiology, Cardiovascular Disease in the Young, and Basic Science, American Heart Association]. Circulation 1998;98:1460–71.
- ↑ Schwartz K, Carrier L, Guicheney P, Komajda M. Molecular basis of familial cardiomyopathies. Circulation 1995;91:532–40.
- ↑ Niimura H, Bachinski LL, Sangwatanaroj S et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 1998;338:1248–57.
- ↑ Thierfelder L, Watkins H, MacRae C et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy. A disease of the sarcomere. Cell 1994;77:701–12.
- ↑ Watkins H, McKenna WJ, Thierfelder L et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 1995;332:1058–64.
- ↑ Charron P, Dubourg O, Desnos M et al. Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin-binding protein C gene. Circulation 1998;97: 2230–6.
- ↑ Maron BJ, Niimura H, Casey SA et al. Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J Am Coll Cardiol 2001;38:315–21.
- ↑ Anan R, Greve G, Thierfelder L et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest 1994;93:280–5.
- ↑ Coviello DA, Maron BJ, Spirito P et al. Clinical features of hypertrophic cardiomyopathy caused by mutation of a “hot spot” in the alpha-tropomyosin gene. J Am Coll Cardiol 1997;29:635–40.
- ↑ Blair E, Redwood C, Ashrafian H et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy. Evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 2001;10:1215–20.
- ↑ Erdmann J, Raible J, Maki-Abadi J et al. Spectrum of clinical phenotypes and gene variants in cardiac myosin-binding protein C mutation carriers with hypertrophic cardiomyopathy. J Am Coll Cardiol 2001;38:322–30.
- ↑ Gruver EJ, Fatkin D, Dodds GA et al. Familial hypertrophic cardiomyopathy and atrial fibrillation caused by Arg663His beta-cardiac myosin heavy chain mutation. Am J Cardiol 1999;83:13H–8H.
- ↑ Kimura A, Harada H, Park JE et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 1997;16:379–82.
- ↑ Marian AJ, Roberts R. Recent advances in the molecular genetics of hypertrophic cardiomyopathy. Circulation 1995;92:1336–47.
- ↑ Niimura H, Patton KK, McKenna WJ et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002;105:446–51.
- ↑ Seidman JG, Seidman CE. The genetic basis for cardiomyopathy. From mutation identification to mechanistic paradigms. Cell 2001; 104:557–67.
- ↑ Arad M, Benson DW, Perez-Atayde AR et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 2002;109:357–62.
- ↑ Doolan G, Nguyen L, Chung J, Ingles J, Semsarian C. Progression of left ventricular hypertrophy and the angiotensin-converting enzyme gene polymorphism in hypertrophic cardiomyopathy. Int J Cardiol. 2004 Aug; 96(2):157–63. (Medline abstract)
- ↑ Marian AJ, Yu QT, Workman R, Greve G, Roberts R. Angiotensin-converting enzyme polymorphism in hypertrophic cardiomyopathy and sudden cardiac death. Lancet. 1993 Oct 30; 342(8879):1085–6. (Medline abstract)
- ↑ Pellikka PA, Oh JK, Bailey KR, Nichols BA, Monahan KH, Tajik AJ. Dynamic intraventricular obstruction during dobutamine stress echocardiography. A new observation. Circulation 1992;86:1429–32.
- ↑ Okeie K, Shimizu M, Yoshio H et al. Left ventricular systolic dysfunction during exercise and dobutamine stress in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2000;36:856–63.
- ↑ 22.0 22.1 Jiang L, Levine RA, King ME, Weyman AE. An integrated mechanism for the systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy based on echocardiographic observations. Am Heart J 1987; 113:633–44
- ↑ 23.0 23.1 Sherrid MV, Gunsburg DZ, Moldenhauer S, Pearle G. Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2000; 36:1344–54
- ↑ Sherrid MV, Chu Ck, DeLia E, Mogtader A, Dwyer Jr. EM, An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22:816–25
- ↑ Levine RA, Vlahakes GJ, Lefebvre X, et al. Papillary muscle displacement causes systolic anterior motion of the mitral valve. Circulation 1995; 91:1189–95
- ↑ Messmer BJ. Extended myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 1994; 58:575–7
- ↑ Schoendube FA, Klues HG, Reith S, Flachskampf FA, Hanrath P, Messmer BJ. Long-term clinical and echocardiographic follow-up after surgical correction of hypertrophic obstructive cardiomyopathy with extended myectomy and reconstruction of the subvalvular mitral apparatus. Circulation 1995; 92:II-122–7
- ↑ Maron MS, Olivotto I, Betocchi S et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348:295–303.
- ↑ Choudhury L, Mahrholdt H, Wagner A et al. Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002;40:2156–64.
- ↑ Maron MS, Olivotto I, Betocchi S et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348:295–303.
- ↑ Kofflard MJ, Ten Cate FJ, van der Lee C, van Domburg RT. Hypertrophic cardiomyopathy in a large community-based population. Clinical outcome and identification of risk factors for sudden cardiac death and clinical deterioration. J Am Coll Cardiol 2003;41:987–93.
- ↑ Maron MS, Olivotto I, Betocchi S et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348:295–303.
- ↑ 33.0 33.1 Lorenzoni R, Gistri R, Cecchi F, Olivotto I, Chiriatti G, Elliott P; et al. (1998). “Coronary vasodilator reserve is impaired in patients with hypertrophic cardiomyopathy and left ventricular dysfunction”. Am Heart J. 136 (6): 972–81. PMID 9842009.
- ↑ 34.0 34.1 Choudhury L, Elliott P, Rimoldi O, Ryan M, Lammertsma AA, Boyd H; et al. (1999). “Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment”. Basic Res Cardiol. 94 (1): 49–59. PMID 10097830.
Differentiating Hypertrophic Cardiomyopathy from Other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Soroush Seifirad, M.D.[2]
Overview
Cardiomyopathy must be differentiated from athlete heart (which is often confused with HCM on echocardiography), hypertrophy due to hypertension or aortic stenosis; as these have common clinical features, including thickened myocardium on imaging and high QRS voltage on EKGs. On the basis increased LV to aortic gradient, hypertrophic cardiomyopathy must be differentiated from sever volume depletion, subaortic stenosis, and valvular aortic stenosis.
Differentiating Hypertrophic cardiomyopathy from other Diseases
Cardiomyopathy must be differentiated from athlete heart (which is often confused with HCM on echocardiography), hypertrophy due to hypertension or aortic stenosis; as these have common clinical features, including thickened myocardium on imaging and high QRS voltage on EKGs. On the basis increased LV to aortic gradient, hypertrophic cardiomyopathy must be differentiated from sever volume depletion, subaortic stenosis, and valvular aortic stenosis.
Differentiating Hypertrophic cardiomyopathy from other diseases on the basis of increased LV to aortic gradient
On the basis increased LV to aortic gradient, hypertrophic cardiomyopathy must be differentiated from sever volume depletion, subaortic stenosis, and valvular aortic stenosis.
Sever volume depletion:
- These patients may develop hyperdynamic ventricular function in an effort to maintain cardiac output in the setting of normal LV systolic function.
- Here is the sequence of events in this setting:
- Hyperdynamic LV function
- Vigorous blood ejection at a higher velocity than normal
- An increased intracavitary gradient
- This may be mistaken for an increased LVOT gradient.
Subaortic stenosis:
- A congenital abnormality typically caused by a thin membrane of tissue in the LVOT
- Fixed subaortic stenosis can usually be distinguished from HCM and valvular AS by echocardiography or invasive cardiac catheterization.
- There is no systolic anterior motion of the mitral valve, and generally the ventricular wall thickness is normal. Nevertheless, chronic long-standing LV hypertension may finally lead to concentric LVH.
- The aortic valve leaflets are usually normal, but in decades these patients may develop aortic valve damage as well.
Valvular aortic stenosis:
- In addition to potentially causing LVH, narrowing of the aortic valve opening can lead to a significant pressure gradient between the LV and the aorta.
- similar to subaortic stenosis, valvular AS can be distinguished from other pathology by echocardiography or invasive cardiac catheterization.
Differentiating Hypertrophic cardiomyopathy from other diseases on the basis of hypertrophy
Cardiomyopathy must be differentiated from athlete heart (which is often confused with HCM on echocardiography), hypertrophy due to hypertension or aortic stenosis; as these have common clinical features, including thickened myocardium on imaging and high QRS voltage on EKGs.
Quite often, HCM can be mistaken for a condition known as athlete’s heart. Both involve the growth of the myocardium, however, the latter generally is not correlated with incidences of SCD. While HCM can be linked to family history, an athlete’s heart arises purely as a function of intense exercise (usually at least an hour a day, every day. Since the body is operating at high training levels, the heart adapts and grows in order to pump blood more efficiently. The stoppage of exercise for three months generally leads to a decrease in wall/septum thickness in those with an athlete’s heart, whereas those with HCM exhibit no decline.
People with an athlete’s heart do not exhibit an abnormally enlarged septum, and the growth of heart muscle at the septum and free ventricular wall is symmetrical. The asymmetrical growth seen in HCM results in a less-dilated left ventricle. This, in turn, leads to a smaller volume of blood leaving the heart with each beat.
| Athlete’s Heart | HCM | |
|---|---|---|
| Septum thickness | <15 mm | >15 mm |
| Symmetry | Yes (for septum and LV wall) | No (septum much thicker |
| Family history | None | Possibly |
| Deconditioning | Reduction within 3 months | None |
Several criteria can be used to distinguish these two entities:
The degree of left ventricular wall thickness
- In athlete’s heart the LVH is symmetric and less than or equal to 12 mm
- Rarely the LV thickness can be 14-16 mm and this makes it difficult to distinguish from HOCM. Athletes who engage in strength training may develop this pattern, athletes who engage in endurance training do not.
- If the degree of thickening is out of proportion to the type and intensity of exercise, this suggests HOCM
The pattern of left ventricular wall thickness
- Athleste’s heart is symmetric
- HOCM is more often asymmetric, but may in some cases be symmetric
The left ventricular cavity size
- HOCM has smaller LV cavitary dimensions
Aortic stenosis must be differentiated from other cardiac or pulmonary causes of dyspnea, weakness, and dizziness. Furthermore, when left ventricular outflow tract obstruction is present, it is critical to identify whether the obstruction is subvalvular, valvular or supravalvular and whether there is hypertrophic cardiomyopathy (HOCM) or not.[1]
Differentiating Aortic Stenosis from other Diseases
Pulmonary Causes of Dyspnea
Aortic stenosis can be differentiated from pulmonary causes of dyspnea by the presence of:
- A narrow pulse pressure
- A harsh late-peaking systolic murmur heard best at the right second intercostal space with radiation to the carotid arteries
- A delayed slow-rising carotid upstroke (pulsus parvus et tardus) [2]
- Signs of heart failure on examination
Aortic Sclerosis
While a murmur may be heard in aortic sclerosis, there is no fusion of the commisures and no significant obstruction to antegrade blood flow across the aortic valve. As a result, the S2 is normal in aortic sclerosis and the carotid upstroke is normal (i.e. pulsus parvus et tardus is absent).[3]
Mitral Regurgitation
The murmur of aortic stenosis is harsh and best heard at the right second intercostal space while the murmur of mitral regurgitation is blowing, soft and best heard at the apex.[4]
Hypertrophic Obstructive Cardiomyopathy
In HOCM the murmur is dynamic and varies with maneuvers. Moreover, there is a bifid or spoke and dome pattern of the carotid upstroke.[5]
Valvular, Subvalvular and Supravalvular Aortic Stenosis
Differentiating Valvular Aortic Stenosis from Subvalvular Aortic Stenosis
Aortic insufficiency is more often present with subvalvular aortic stenosis (in 50% to 75% of cases). Symptoms associated with subvalvular aortic stenosis begin earlier in life (in childhood or adolescence) than symptoms associated with valvular aortic stenosis.[6]
Differentiating Valvular Aortic Stenosis from Supravalvular Aortic Stenosis
Supravalvular aortic stenosis is an uncommon congenital anomaly caused by a narrowing in the ascending aorta or by the presence of a fibrous diaphragm just above the aortic valve. It presents in early adulthood. Although the aortic valve is not stenotic, doppler shows an increased pressure gradient. 50% of patients with supravalvular aortic stenosis have a characteristically greater pulse and systolic blood pressure in the right carotid and brachial arteries than in the left. The systolic murmur is maximal below the right clavicle and radiates primarily to the right carotid artery. There is not an ejection click nor a diastolic murmur.[6]
Differentiating hypertrophic cardiomyopathy and valvular aortic stenosis
| Diseases | Clinical manifestations | Para-clinical findings | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| History and Physical examination | ||||||||||||
| Lab Findings | Echocardiography | Histopathology | ||||||||||
| Murmur of AI | Pulse pressure after PVC | Valsalva maneuver | Carotid pulsation | Family history | Deconditioning | ECG | Gene mutations | Aortic valve calcification | Dilated ascending aorta | Ventricular hypertrophy | ||
| Hypertrophic cardiomyopathy | No | Decreased | Increased intensity of murmur | Brisk, jerky, or bisferiens pulse (a collapse of the pulse followed by a secondary rise) | + | None |
|
Yes | No | Rare |
|
Myocardial disarray |
| Aortic stenosis | Common | Increased | Decreased intensity of murmur | Normal or tardus et parvus | +/- | None |
|
No | Common | Common | Concentric LVH | Hyperthrophy |
| Athlete’s Heart | No | No | No | Normal | – | Reduction within 3 months |
|
No | No | No |
|
Hyperthrophy |
*A 12 lead EKG is strongly recommended at the time of the initial diagnosis of hypertrophic cardiomyopathy. Common findings on an EKG in these patients include tall R waves, deep Q waves, inverted T waves, ST segment abnormalities and ‘strain pattern’ in the chest leads. The deep Q waves indicate septal hypertrophy and similarly deeply inverted T waves indicate apical hypertrophy.
Other differential diagnoses
- Long-standing HTN is the most common cause of LVH
- Most of the LVH due to HTN cases will present beyond adolescence when HCM is most commonly identified.
- The magnitude of hypertrophy seen in hypertension, however, rarely leads to wall thicknesses in excess of 1.5 cm.
- Hypertension is usually suspected in persons with an extended history of elevated blood pressures (10 or more years), particularly in those with other evidence of end-organ damage due to hypertension (eg, retinopathy, nephropathy).
(X-linked deficiency of the lysosomal enzyme alphagalactosidase)
References
- ↑ Cleland JG, Swedberg K, Follath F, Komajda M, Cohen-Solal A, Aguilar JC, Dietz R, Gavazzi A, Hobbs R, Korewicki J, Madeira HC, Moiseyev VS, Preda I, van Gilst WH, Widimsky J, Freemantle N, Eastaugh J, Mason J (2003). “The EuroHeart Failure survey programme– a survey on the quality of care among patients with heart failure in Europe. Part 1: patient characteristics and diagnosis”. European Heart Journal. 24 (5): 442–63. PMID 12633546. Retrieved 2012-04-11. Unknown parameter
|month=ignored (help) - ↑ Toy, Eugene, et al. Case Files: Internal Medicine. McGraw-Hill Companies, Inc. 2007. Page 43. ISBN 0071463038.
- ↑ Lucena CM, Santos RP (2015). “Association between Aortic Valve Sclerosis and Adverse Cardiovascular Events”. Arq Bras Cardiol. 105 (1): 99. doi:10.5935/abc.20150081. PMC 4523295. PMID 26270071.
- ↑ Mirabel M, Iung B, Baron G, Messika-Zeitoun D, Détaint D, Vanoverschelde JL; et al. (2007). “What are the characteristics of patients with severe, symptomatic, mitral regurgitation who are denied surgery?”. Eur Heart J. 28 (11): 1358–65. doi:10.1093/eurheartj/ehm001. PMID 17350971.
- ↑ Veselka J, Anavekar NS, Charron P (2016). “Hypertrophic obstructive cardiomyopathy”. Lancet. doi:10.1016/S0140-6736(16)31321-6. PMID 27912983.
- ↑ 6.0 6.1 Roberts WC (1973). “Valvular, subvalvular and supravalvular aortic stenosis: morphologic features”. Cardiovasc Clin. 5 (1): 97–126. PMID 4272665.
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Ogheneochuko Ajari, MB.BS, MS [2] Soroush Seifirad, M.D.[3]
Overview
Hypertrophic cardiomyopathy is a condition that is most often passed down through families (inherited). It is thought to result from gene mutations that control heart muscle growth. Genes involved in the pathogenesis of hypertrophic cardiomyopathy include MYH7, TNNT2, TPM1. Nevertheless, a number of chronic medical conditions might be contributed to hypertrophic cardiomyopathy development, among them are thyroid disease, diabetes, and obesity, and hypertension.
Causes
Life Threatening Causes
Life-threatening causes include conditions that may result in death or permanent disability within 24 hours if left untreated.
Common Causes
- Familial
- Gene mutation
- Hypertension
- Thyroid disease
- Diabetes
- Obesity
Causes by Organ System
Causes in Alphabetical Order
- Aging
- Atrial myxoma[1]
- Cardiofaciocutaneous syndrome
- Congenital generalized lipodystrophy type 2
- Costello syndrome [2]
- Cytochrome c oxidase deficiency
- Diabetes mellitus
- Dihydrolipoamide dehydrogenase deficiency
- Fabry’s disease[3]
- Familial
- Friedreich’s ataxia[4]
- Gene mutation
- Glycogenosis type 2
- Hereditary spherocytosis
- Hypertension
- Hypertrichotic osteochondrodysplasia
- Hypertrophic obstructive cardiomyopathy
- Idiopathic
- Long-chain acyl-CoA dehydrogenase deficiency
- Malonyl-CoA decarboxylase deficiency
- MELAS
- Multiple lentigines syndrome
- Muscle glycogen synthase deficiency
- Myotonic dystrophy[5]
- Noonan syndrome[6]
- Sarcomeric protein mutations
- Subendocardial ischemia
- Thyroid disease
- Very long-chain acyl-CoA dehydrogenase deficiency[7]
- Yunis-Varon syndrome
Genetics
Hypertrophic cardiomyopathy is transmitted in an autosomal dominant pattern.
Genes involved in the pathogenesis of hypertrophic cardiomyopathy include:
The development of Hypertrophic cardiomyopathy is the result of multiple genetic mutations such as:
- Beta-myosin heavy chain
- Myosin binding protein C
- Cardiac troponin T
HCM is the most common genetically transmitted cardiovascular disease. Hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins. Penetrance of HCM is incomplete, variable and time or age-related. The disease may be sporadic but affected family members are discovered in 13% of cases. More than 200 mutations involving at least 10 chromosomes encoding structural proteins of the myocyte have been discovered. These mutations have varying degrees of penetrance and even the same mutation may have variable expression, implying the superimposed effects of other genes or environmental influences. Children of a patient with HCM have a 50% chance of inheriting the trait.
References
- ↑ Abdou M, Hayek S, Williams BR (2013). “Atrial myxoma in a patient with hypertrophic cardiomyopathy”. Tex Heart Inst J. 40 (4): 462–4. PMC 3783132. PMID 24082380.
- ↑ Fukao T, Sakai S, Shimozawa N, Kuwahara T, Kano M, Goto E; et al. (1996). “Life-threatening cardiac involvement throughout life in a case of Costello syndrome”. Clin Genet. 50 (4): 244–7. PMID 9001809.
- ↑ Adam T, Alexandrescu L, Voinea F, Toringhibel M, Hâncu A (2006). “Fabry’s disease”. Rom J Intern Med. 44 (4): 455–64. PMID 18386622.
- ↑ Jauslin ML, Wirth T, Meier T, Schoumacher F (2002). “A cellular model for Friedreich Ataxia reveals small-molecule glutathione peroxidase mimetics as novel treatment strategy”. Hum Mol Genet. 11 (24): 3055–63. PMID 12417527.
- ↑ Halawa A, Iskandar SB, Brahmbhatt V, Fahrig SA (2007). “Atrial flutter and myotonic dystrophy in a male adolescent treated with radiofrequency catheter ablation”. Rev Cardiovasc Med. 8 (2): 118–22. PMID 17603429.
- ↑ Prendiville TW, Gauvreau K, Tworog-Dube E, Patkin L, Kucherlapati RS, Roberts AE; et al. (2014). “Cardiovascular disease in Noonan syndrome”. Arch Dis Child. 99 (7): 629–34. doi:10.1136/archdischild-2013-305047. PMID 24534818.
- ↑ Tucci S, Flögel U, Hermann S, Sturm M, Schäfers M, Spiekerkoetter U (2014). “Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD(-/-)) mice”. Biochim Biophys Acta. 1842 (5): 677–85. doi:10.1016/j.bbadis.2014.02.001. PMID 24530811.
Epidemiology and Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Soroush Seifirad, M.D.[2]
Overview
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease. Prevalence rates have been reported between 1:500 (0.2%) and 1:3,000 (0.03%) because of variations in study designs and cohort characteristics including different age groups and ethnicity. According to the CARDIA (Coronary Artery Risk Development in Young Adults) cohort study that used standard echocardiography in 4,111 unrelated people 23 to 35 years of age, HCM prevalence is reported as 1 in 500 persons (0.2%). Nevertheless, lower prevalence has been reported in some European countries such as Germany (0.07%). Patients of all age groups may develop hypertrophic cardiomyopathy. Prevalence increased with advancing age and showed a constant yearly rise but sudden death is more prevalent in young patients, particularly athletes. The case-fatality rate is 6 per 10,000 per year in young people without symptoms of hypertrophic cardiomyopathy but in syptomatic patients a case-fatality rate is 420 and 110 deaths per 10,000 per year in tertiary referral centers and general hospital clinics respectively. Hypertrophic cardiomyopathy affects men and women equally. However, despite more frequent outflow obstruction, women with HCM are underrecognized and referred to centers later than men, often with more advanced heart failure. Greater awareness of HCM in women should lead to earlier diagnosis and treatment, with implications for improved quality of life. HCM is less prevalent in African Americans, but they are more pron to early presentation, developing heart failure, and sudden death is more prevalent due to less awareness and screening in this population.
Epidemiology and Demographics
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease. Prevalence rates have been reported between 1:500 (0.2%) and 1:3,000 (0.03%) because of variations in study designs and cohort characteristics including different age groups and ethnicity. According to the CARDIA (Coronary Artery Risk Development in Young Adults) cohort study that used standard echocardiography in 4,111 unrelated people 23 to 35 years of age, HCM prevalence is reported as 1 in 500 persons (0.2%).[1] Nevertheless, lower prevalence has been reported in some European countries such as Germany (0.07%). [2]A recent analysis of U.S. claims data reported a prevalence of clinically diagnosed HCM in approximately 1:3,000 (0.03%)[3] According to Semsarian et al. “For the past 20 years, most data have supported the occurrence of HCM at about 1 in 500. However, the authors have interrogated a number of relevant advances in cardiovascular medicine, including widespread fee-for-service genetic testing, population genetic studies, and contemporary diagnostic imaging, as well as a greater index of suspicion and recognition for both the clinically expressed disease and the gene-positive– phenotype-negative subset (at risk for developing the disease). Accounting for the potential impact of these initiatives on disease occurrence, the authors have revisited the prevalence of HCM in the general population.”[4]
Incidence
- The incidence/prevalence of Hypertrophic cardiomyopathy is approximately [number range] per 100,000 individuals worldwide.
- In [year], the incidence/prevalence of Hypertrophic cardiomyopathy was estimated to be [number range] cases per 100,000 individuals worldwide.
Prevalence
- The prevalence of hypertrophic cardiomyopathy is approximately 200 per 100,000 individuals worldwide. Nevertheless recent studies suggested a higher prevalence. [4]
Case-fatality rate/Mortality rate
- The case-fatality rate/mortality rate of is different in asymptomatic vs symptomatic patients.
- The case-fatality rate is 6 per 10,000 per year in young people without symptoms of hypertrophic cardiomyopathy but in syptomatic patients a case-fatality rate is 420 and 110 deaths per 10,000 per year in tertiary referral centers and general hospital clinics respectively.
- Although it is quite prevalent, hypertrophic cardiomyopathy rarely causes death; the case-fatality rate is about 6 per 10,000 per year in young people without symptoms of hypertrophic cardiomyopathy.[5]
- Current risk estimates from the study of patients in tertiary referral centers or general hospital clinics are not applicable to asymptomatic people in the general population.[6]
Age
- Patients of all age groups may develop hypertrophic cardiomyopathy.
- Patients can be diagnosed at any age, from birth to age 80 and beyond.
- Children and adolescents with the condition usually come to attention when a family screening is performed after an adult in the family is found to be affected or at the presence of a heart murmur that is evaluated more closely.
- Approximately 50% of adults with the condition present with symptoms, the average age of diagnosis within the HCMA database is 39 years.
- Prevalence increased with advancing age and showed a constant yearly rise.
- But sudden death is more prevalent in young patients, particularly athletes.
- Hypertrophic cardiomyopathy can cause heart-related sudden death in people of all ages, but the condition most often causes sudden cardiac death in people under the age of 30.
Race
- For years, HCM was considered an uncommon condition among the African American population. Maron et al, in a multicenter analysis of clinically diagnosed HCM patients noted only 8% of the study population was black. In Movahed et al study, African Americans constitute only 9% of the population. However, in the light of several premature cardiovascular deaths among African American athletes, Maron et al, in an autopsy analysis of young athletes surprisingly noted HCM was 7 times more commonly identified in African Americans for the first time at autopsy compared to when clinically identified in African American population.[7]
- HCM-related sudden death is more prevalent in black male athletes.[8]Many HCM cases go unrecognized in the African American community, underscoring the need for enhanced clinical recognition of HCM to create the opportunity for preventive measures to be employed in high-risk patients with this complex disease.
- According to Sheikh et al who studied 425 patients in the UK, there are differences in presenting features including the prevalence of hypertension and an abnormal ECG in black and white patients with HCM. Apical and concentric hypertrophy is more common in black patients and may have implications for appropriate diagnosis, especially in those with concurrent hypertension. Overall freedom from adverse events is similar.[9]
- Sorensen et al compared 76 blacks for clinical presentation, electrocardiogram, exercise capacity, left ventricular morphology, and hemodynamics by echocardiography to 365 whites. They concluded that “blacks have an HC phenotype characterized by a lower prevalence of the well-recognized echocardiographic features of HC such as the systolic anterior movement of the mitral valve and left ventricular outflow tract obstruction and display worse exercise capacity.”[10]
- The Sarcomeric Human Cardiomyopathy Registry (1989 – 2018)
- Lauren A. Eberly, M.D., identified 2,467 patients with hypertrophic cardiomyopathy (8.3 percent black; 91.7 percent white). Black patients were younger at the time of diagnosis (mean age, 36.5 versus 41.9 years), had a higher prevalence of New York Heart Association (NYHA) class III or IV heart failure at presentation (22.6 versus 15.8 percent), had lower rates of genetic testing (54.1 versus 62.1 percent) and were less likely to have sarcomeric mutations identified by genetic testing (26.1 versus 40.5 percent) compared with white patients. There were no racial differences noted in implantation of implantable cardioverter-defibrillators, but the invasive septal reduction was less common among black patients (14.6 versus 23 percent). Black patients had less incident atrial fibrillation (35 [17.1 percent] versus 608 [26.9 percent]). There was an association between black race and increased development of NYHA class III or IV heart failure (hazard ratio, 1.45).[11]
- The United States National Registry
- Within this large forensic registry of competitive athletes, cardiovascular sudden deaths due to genetic and/or congenital heart diseases were uncommon in females and more common in African Americans/other minorities than in whites. Hypertrophic cardiomyopathy is an under-appreciated cause of sudden death in male minority athletes.[12]
Germany
- Prevalence of clinically diagnosed HCM in Germany is lower than in systematic population studies based on the echocardiographic diagnosis.
Gender
- Hypertrophic cardiomyopathy affects men and women equally.
- Survival was not less favorable in women with HCM.
- Contemporary treatments including surgical myectomy to reverse heart failure and defibrillators to prevent sudden death, were effective in both sexes contributing to low mortality.
- However, despite more frequent outflow obstruction, women with HCM are underrecognized and referred to centers later than men, often with more advanced heart failure. Greater awareness of HCM in women should lead to earlier diagnosis and treatment, with implications for improved quality of life.[13][14]
Region
- The majority of Hypertrophic cardiomyopathy cases are reported in [geographical region].
- Hypertrophic cardiomyopathy is a common/rare disease that tends to affect [patient population 1] and [patient population 2].
Developed Countries
Developing Countries
References
- ↑ Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE (1995). “Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults”. Circulation. 92 (4): 785–9. doi:10.1161/01.cir.92.4.785. PMID 7641357.
- ↑ Husser D, Ueberham L, Jacob J, Heuer D, Riedel-Heller S, Walker J; et al. (2018). “Prevalence of clinically apparent hypertrophic cardiomyopathy in Germany-An analysis of over 5 million patients”. PLoS One. 13 (5): e0196612. doi:10.1371/journal.pone.0196612. PMC 5933727. PMID 29723226.
- ↑ Maron MS, Hellawell JL, Lucove JC, Farzaneh-Far R, Olivotto I (2016). “Occurrence of Clinically Diagnosed Hypertrophic Cardiomyopathy in the United States”. Am J Cardiol. 117 (10): 1651–1654. doi:10.1016/j.amjcard.2016.02.044. PMID 27006153.
- ↑ 4.0 4.1 Semsarian C, Ingles J, Maron MS, Maron BJ (2015). “New perspectives on the prevalence of hypertrophic cardiomyopathy”. J Am Coll Cardiol. 65 (12): 1249–1254. doi:10.1016/j.jacc.2015.01.019. PMID 25814232.
- ↑ Wald DS, Law M, Morris JK (2004). “Mortality from hypertrophic cardiomyopathy in England and Wales: clinical and screening implications”. Int J Cardiol. 97 (3): 479–84. doi:10.1016/j.ijcard.2003.11.014. PMID 15561336.
- ↑ Maron BJ, Rowin EJ, Casey SA, Maron MS (2016). “How Hypertrophic Cardiomyopathy Became a Contemporary Treatable Genetic Disease With Low Mortality: Shaped by 50 Years of Clinical Research and Practice”. JAMA Cardiol. 1 (1): 98–105. doi:10.1001/jamacardio.2015.0354. PMID 27437663.
- ↑ Movahed MR, Strootman D, Bates S, Sattur S (2010). “Prevalence of suspected hypertrophic cardiomyopathy or left ventricular hypertrophy based on race and gender in teenagers using screening echocardiography”. Cardiovasc Ultrasound. 8: 54. doi:10.1186/1476-7120-8-54. PMC 3019148. PMID 21143986.
- ↑ Maron BJ, Carney KP, Lever HM, Lewis JF, Barac I, Casey SA; et al. (2003). “Relationship of race to sudden cardiac death in competitive athletes with hypertrophic cardiomyopathy”. J Am Coll Cardiol. 41 (6): 974–80. doi:10.1016/s0735-1097(02)02976-5. PMID 12651044.
- ↑ Sheikh N, Papadakis M, Panoulas VF, Prakash K, Millar L, Adami P; et al. (2016). “Comparison of hypertrophic cardiomyopathy in Afro-Caribbean versus white patients in the UK”. Heart. 102 (22): 1797–1804. doi:10.1136/heartjnl-2016-309843. PMID 27679836.
- ↑ Sorensen LL, Pinheiro A, Dimaano VL, Pozios I, Nowbar A, Liu H; et al. (2016). “Comparison of Clinical Features in Blacks Versus Whites With Hypertrophic Cardiomyopathy”. Am J Cardiol. 117 (11): 1815–20. doi:10.1016/j.amjcard.2016.03.017. PMID 27084053.
- ↑ Eberly LA, Day SM, Ashley EA, Jacoby DL, Jefferies JL, Colan SD; et al. (2019). “Association of Race With Disease Expression and Clinical Outcomes Among Patients With Hypertrophic Cardiomyopathy”. JAMA Cardiol. doi:10.1001/jamacardio.2019.4638. PMID 31799990.
- ↑ Maron BJ, Haas TS, Ahluwalia A, Murphy CJ, Garberich RF (2016). “Demographics and Epidemiology of Sudden Deaths in Young Competitive Athletes: From the United States National Registry”. Am J Med. 129 (11): 1170–1177. doi:10.1016/j.amjmed.2016.02.031. PMID 27039955.
- ↑ Rowin EJ, Maron MS, Wells S, Patel PP, Koethe BC, Maron BJ (2019). “Impact of Sex on Clinical Course and Survival in the Contemporary Treatment Era for Hypertrophic Cardiomyopathy”. J Am Heart Assoc. 8 (21): e012041. doi:10.1161/JAHA.119.012041. PMC 6898820 Check
|pmc=value (help). PMID 31663408. - ↑ Maron BJ (2018). “Clinical Course and Management of Hypertrophic Cardiomyopathy”. N Engl J Med. 379 (7): 655–668. doi:10.1056/NEJMra1710575. PMID 30110588.
References
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Soroush Seifirad, M.D.[2]
Overview
Obstructive hypertrophic cardiomyopathy (HCOM) is known as a familial genetic disorder. The most potent risk factor in the development of hypertrophic cardiomyopathy aregenetic mutations in Beta-myosin heavy chain, Myosin binding protein C, and Cardiac troponin T. Genes involved in the pathogenesis of hypertrophic cardiomyopathy include but not limited to MYH7, TNNT2, TPM1. However, hypertension, thyroid disease, diabetes, and obesity also play a role in non obstructive forms of hypertrophic cardiomyopathy. This is in response to chronic effects of abnormal pressure and volumes on the myocardium and is different from apical hypertrophy (Yamaguchi syndrome).
Risk Factors
Obstructive hypertrophic cardiomyopathy (HCOM) is known as a familial genetic disorder. The most potent risk factor in the development of hypertrophic cardiomyopathy aregenetic mutations in Beta-myosin heavy chain, Myosin binding protein C, and Cardiac troponin T. Genes involved in the pathogenesis of hypertrophic cardiomyopathy include but not limited to MYH7, TNNT2, TPM1. However, hypertension, thyroid disease, diabetes, and obesity also play a role in non obstructive forms of hypertrophic cardiomyopathy. This is in response to chronic effects of abnormal pressure and volumes on the myocardium and is different from apical hypertrophy (Yamaguchi syndrome).
Common Risk Factors
- Common risk factors in the development of hypertrophic cardiomyopathy are genetic mutations in Beta-myosin heavy chain, Myosin binding protein C, and Cardiac troponin T.
- Genes involved in the pathogenesis of hypertrophic cardiomyopathy include:
Less Common Risk Factors (risk factors for non obstructive/non genetic forms of disease)
- Less common risk factors in the development of non obstructive/non genetic hypertrophic cardiomyopathy include:
Risk Factors for Sudden Cardiac Death
Patients with hypertrophic cardiomyopathy are increased risk of sudden cardiac death. Identification of those patients at an increased risk can facilitate early treatment with an automatic implantable cardiac defibrillator.
Risk Factors for Sudden Cardiac Death in Patients with Hypertrophic Cardiomyopathy
Risk factors for sudden death in individuals with HCM include:[1]
- A young age at first diagnosis (age < 30 years)
- An episode of aborted sudden death
- A family history of HCM with sudden death of relatives
- Specific mutations in the genes encoding for troponin T and myosin
- Sustained supraventricular or ventricular tachycardia
- Ventricular septal wall thickness over 3 cm
- A hypotensive response to exercise
- Recurrent syncope (especially in children)
- Bradyarrhythmias (slow rhythms of the heart).
2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy (DO NOT EDIT)[2]
SCD Risk Stratification (DO NOT EDIT)[2]
| Class I |
| “1. All patients with HCM should undergo comprehensive SCD risk stratification at initial evaluation to determine the presence of the following:[3][4][5][6][7][8][9][10][11][12][13][14] (Level of Evidence: B)“ a. A personal history for ventricular fibrillation, sustained VT, or SCD events, including appropriate ICD therapy for ventricular tachyarrhythmias b. A family history for SCD events, including appropriate ICD therapy for ventricular tachyarrhythmias. c. Unexplained syncope. d. Documented NSVT defined as 3 or more beats at greater than or equal to 120 bpm on ambulatory (Holter) ECG. e. Maximal LV wall thickness greater than or equal to 30 mm. |
| Class IIa |
| “1. It is reasonable to assess blood pressure response during exercise as part of SCD risk stratification in patients with HCM[3][15][11]. (Level of Evidence: B)” |
| “2. SCD risk stratification is reasonable on a periodic basis (every 12 to 24 months) for patients with HCM who have not undergone ICD implantation but would otherwise be eligible in the event that risk factors are identified (12 to 24 months). (Level of Evidence: C)” |
| Class IIb |
| “1. The usefulness of the following potential SCD risk modifiers is unclear but might be considered in selected patients with HCM for whom risk remains borderline after documentation of conventional risk factors:” |
| “a. CMR imaging with LGE[16][17]. (Level of Evidence: C)” |
| “b. Double and compound mutations (ie, >1). (Level of Evidence: C)” |
| “c. Marked LVOT obstruction[3][18][19][11]. (Level of Evidence: B)” |
| Class III |
| “1. Invasive electrophysiologic testing as routine SCD risk stratification for patients with HCM should not be performed. (Level of Evidence: C)” |
References
- ↑ Maron BJ, Cecchi F, McKenna WJ (1994). “Risk factors and stratification for sudden cardiac death in patients with hypertrophic cardiomyopathy”. Br Heart J. 72 (6 Suppl): S13–8. doi:10.1136/hrt.72.6_Suppl.S13. PMC 1025670. PMID 7873317. Unknown parameter
|month=ignored (help) and the Task Force on Sudden Cardiac Death of the European Society of Cardiology link Note: Guideline withdraw - ↑ 2.0 2.1 Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW (2011). “2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons”. Journal of the American College of Cardiology. 58 (25): e212–60. doi:10.1016/j.jacc.2011.06.011. PMID 22075469. Retrieved 2011-12-19. Unknown parameter
|month=ignored (help) - ↑ 3.0 3.1 3.2 Elliott PM, Gimeno JR, Tomé MT; et al. (2006). “Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy”. Eur. Heart J. 27 (16): 1933–41. doi:10.1093/eurheartj/ehl041. PMID 16754630. Unknown parameter
|month=ignored (help) - ↑ Maron BJ, Savage DD, Wolfson JK, Epstein SE (1981). “Prognostic significance of 24 hour ambulatory electrocardiographic monitoring in patients with hypertrophic cardiomyopathy: a prospective study”. Am. J. Cardiol. 48 (2): 252–7. PMID 7196685. Unknown parameter
|month=ignored (help) - ↑ Maron BJ (2010). “Risk stratification and role of implantable defibrillators for prevention of sudden death in patients with hypertrophic cardiomyopathy”. Circ. J. 74 (11): 2271–82. PMID 20962423. Unknown parameter
|month=ignored (help) - ↑ Maron BJ (2010). “Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy”. Circulation. 121 (3): 445–56. doi:10.1161/CIRCULATIONAHA.109.878579. PMID 20100987. Unknown parameter
|month=ignored (help) - ↑ Cecchi F, Maron BJ, Epstein SE (1989). “Long-term outcome of patients with hypertrophic cardiomyopathy successfully resuscitated after cardiac arrest”. J. Am. Coll. Cardiol. 13 (6): 1283–8. PMID 2703610. Unknown parameter
|month=ignored (help) - ↑ Elliott PM, Sharma S, Varnava A, Poloniecki J, Rowland E, McKenna WJ (1999). “Survival after cardiac arrest or sustained ventricular tachycardia in patients with hypertrophic cardiomyopathy”. J. Am. Coll. Cardiol. 33 (6): 1596–601. PMID 10334430. Unknown parameter
|month=ignored (help) - ↑ Elliott PM, Poloniecki J, Dickie S; et al. (2000). “Sudden death in hypertrophic cardiomyopathy: identification of high risk patients”. J. Am. Coll. Cardiol. 36 (7): 2212–8. PMID 11127463. Unknown parameter
|month=ignored (help) - ↑ Fananapazir L, Chang AC, Epstein SE, McAreavey D (1992). “Prognostic determinants in hypertrophic cardiomyopathy. Prospective evaluation of a therapeutic strategy based on clinical, Holter, hemodynamic, and electrophysiological findings”. Circulation. 86 (3): 730–40. PMID 1516184. Unknown parameter
|month=ignored (help) - ↑ 11.0 11.1 11.2 Maki S, Ikeda H, Muro A; et al. (1998). “Predictors of sudden cardiac death in hypertrophic cardiomyopathy”. Am. J. Cardiol. 82 (6): 774–8. PMID 9761089. Unknown parameter
|month=ignored (help) - ↑ Maron BJ, Spirito P, Shen WK; et al. (2007). “Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy”. JAMA. 298 (4): 405–12. doi:10.1001/jama.298.4.405. PMID 17652294. Unknown parameter
|month=ignored (help) - ↑ McKenna W, Deanfield J, Faruqui A, England D, Oakley C, Goodwin J (1981). “Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features”. Am. J. Cardiol. 47 (3): 532–8. PMID 7193406. Unknown parameter
|month=ignored (help) - ↑ Spirito P, Autore C, Rapezzi C; et al. (2009). “Syncope and risk of sudden death in hypertrophic cardiomyopathy”. Circulation. 119 (13): 1703–10. doi:10.1161/CIRCULATIONAHA.108.798314. PMID 19307481. Unknown parameter
|month=ignored (help) - ↑ Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ (1997). “Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy”. Circulation. 96 (9): 2987–91. PMID 9386166. Unknown parameter
|month=ignored (help) - ↑ Adabag AS, Maron BJ, Appelbaum E; et al. (2008). “Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance”. J. Am. Coll. Cardiol. 51 (14): 1369–74. doi:10.1016/j.jacc.2007.11.071. PMID 18387438. Unknown parameter
|month=ignored (help) - ↑ Moon JC, McKenna WJ, McCrohon JA, Elliott PM, Smith GC, Pennell DJ (2003). “Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance”. J. Am. Coll. Cardiol. 41 (9): 1561–7. PMID 12742298. Unknown parameter
|month=ignored (help) - ↑ Efthimiadis GK, Parcharidou DG, Giannakoulas G; et al. (2009). “Left ventricular outflow tract obstruction as a risk factor for sudden cardiac death in hypertrophic cardiomyopathy”. Am. J. Cardiol. 104 (5): 695–9. doi:10.1016/j.amjcard.2009.04.039. PMID 19699347. Unknown parameter
|month=ignored (help) - ↑ Maron MS, Olivotto I, Betocchi S; et al. (2003). “Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy”. N. Engl. J. Med. 348 (4): 295–303. doi:10.1056/NEJMoa021332. PMID 12540642. Unknown parameter
|month=ignored (help)
Screening
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-In-Chief: Lakshmi Gopalakrishnan, M.B.B.S. [2] Soroush Seifirad, M.D.[3]
Overview
Genetic testing is the diagnostic study of choice to definitively diagnose hypertrophic cardiomyopathy. While definitive, these techniques can be expensive and can be difficult to access. If the mutation has already been identified in other family members, it is fairly efficient to test for that isolated mutation. Once HCM has been identified in a family, immediate testing of all family members will help to identify those at risk.
Screening Methods if Genetic Testing is Not Available
Absent the availability of genetic testing, clinical screening should be conducted using the history, physical exam, the electrocardiogram and the echocardiogram in adolescent patients aged 12 to 18 who are first degree relatives of patients with a confirmed diagnosis of hypertrophic cardiomyopathy. Because HCM can have a delayed age of onset, individuals over the age of 18 with an affected first degree relative should have screening every 5 years. Unless the child is engaged in extremely competitive sports or has an aggressive family history of HCM with premature death, screening is generally not recommended in children under the age of 12.
Screening of Competitive Athletes for Hypertrophic Cardiomyopathy
Screening for hypertrophic cardiomyopathy(HCM) is a controversial subject in the medical community, as HCM is the leading cause of sudden death in athletes.
Family History
The AHA/ACC guidelines recommend that a family history should be obtained in athletes to ascertain if there is a history of sudden death or if HCM is present in any family members.
Physical Examination
If the family history is positive, then a physical examination and echocardiography should be performed.
Yield of Aggressive Testing: The Italian Experience
- In Italy, all competitive athletes are required to undergo pre-participation screening for the presence of HCM.
- This screening consists of:
- A 12-lead ECG
- A general and cardiovascular physical examination, including blood pressure measurements
- A family history
- Over 3 million competitive athletes are evaluated each year, and athletes judged to be free of cardiovascular disease receive a certificate enabling them to participate in competitive athletics.
- In a study done in Italy over a 9-year period (1990-1998), 41 of the 4450 athletes screened on echocardiography showed LV hypertrophy, with an increased wall thicknesses. Only four athletes demonstrated a maximal LV wall thickness of 13 mm or more. Thus, the yield of echocardiographic screening appears to be low.
Follow-Up of Patients with diagnosed HOCM
These patients are re-evaluated every 12 to 18 months.
2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines[1]
Recommendations for Genetics and Family Screening Referenced studies that support the recommendations are summarized in the Online Data Supplement
| Class I |
| 1. In patients with HCM, evaluation of familial inheritance, including a 3-generation family history, is recommended as part of the initial assessment.(Level of Evidence: B-NR)
2. In patients with HCM, genetic testing is beneficial to elucidate the genetic basis to facilitate the identification of family members at risk for developing HCM (cascade testing)(Level of Evidence: B-NR) 3. In patients with an atypical clinical presentation of HCM or when another genetic condition is suspected to be the cause, a work-up including genetic testing for HCM and other genetic causes of unexplained cardiac hypertrophy (“HCM phenocopies”) is recommended(Level of Evidence: B-NR) 4. In patients with HCM who choose to undergo genetic testing, pre- and posttest genetic counseling by an expert in the genetics of cardio-vascular disease is recommended so that risks, benefits, results, and their clinical significance can be reviewed and discussed with the patient in a shared decision-making process.(Level of Evidence: B-NR) 5. When performing genetic testing in an HCM proband, the initial tier of genes tested should include genes with strong evidence to be disease-causing in HCM(Level of Evidence: B-NR) 6. In first-degree relatives of patients with HCM, both clinical screening (ECG and 2D echocardiogram) and cascade genetic testing (when a pathogenic/likely pathogenic variant has been identified in the proband) should be offered.(Level of Evidence: B-NR) 7. In families where a sudden unexplained death has occurred with a postmortem diagnosis of HCM, postmortem genetic testing is beneficial to facilitate cascade genetic testing and clinical screening in first-degree relatives.(Level of Evidence: B-NR) 8. In patients with HCM who have undergone genetic testing, serial reevaluation of the clinical significance of the variant(s) identified is recommended to assess for variant reclassification, which may impact diagnosis and cascade genetic testing in family members(Level of Evidence: B-NR) 9. In affected families with HCM, preconception and prenatal reproductive and genetic counseling should be offered(Level of Evidence: B-NR) |
| Class IIb |
| 10. In patients with HCM, the usefulness of genetic testing in the assessment of risk of SCD is uncertain(Level of Evidence: B-NR)
1. In patients with HCM who harbor a variant of uncertain significance, the usefulness of clinical genetic testing of phenotype-negative relatives for the purpose of variant reclassification is uncertain.(Level of Evidence: B-NR) |
Recommendations for Individuals Who Are Genotype-Positive, Phenotype-Negative Referenced studies that support the recommendations are summarized in the Online Data Supplement
| Class I |
| 1. In individuals who are genotype-positive, phenotype-negative for HCM, serial clinical assessment, electrocardiography, and cardiac imaging are recommended at periodic intervals depending on age (every 1 to 2 years in children and adolescents, and every 3 to 5 years in adults) and change in clinical status1–5(Figure 1 and Figure 2, Table 6)(Level of Evidence: B-NR) |
| Class IIa |
| 2. In individuals who are genotype-positive, phenotype-negative for HCM, participation in competitive athletics of any intensity is reasonable(Level of Evidence: C-LD) |
2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy (DO NOT EDIT)[2]
Genetic Testing Strategies/Family Screening (DO NOT EDIT) [2]
| Class I |
| ”1. Evaluation of familial inheritance and genetic counseling is recommended as part of the assessment of patients with HCM.[3][4][5][6][7][8] (Level of Evidence: B)” |
| ”2. Patients who undergo genetic testing should also undergo counseling by someone knowledgeable in the genetics of cardiovascular disease so that results and their clinical significance can be appropriately reviewed with the patient.[9][10][11][12][13] (Level of Evidence: B)” |
| ”3. Screening (clinical, with or without genetic testing) is recommended in first-degree relatives of patients with HCM.[3][4][5][6][8][14][15] (Level of Evidence: B)” |
| ”4. Genetic testing for HCM and other genetic causes of unexplained cardiac hypertrophy is recommended in patients with an atypical clinical presentation of HCM or when another genetic condition is suspected to be the cause.[16][17][18] (Level of Evidence: B)” |
| Class IIa |
| ”1. Genetic testing is reasonable in the index patient to facilitate the identification of first-degree family members at risk for developing HCM.[3][7][14] (Level of Evidence: B)” |
| Class IIb |
| ”1. The usefulness of genetic testing in the assessment of risk of SCD in HCM is uncertain.[19][20] (Level of Evidence: B)” |
| Class III (No Benefit) |
| ”1. Genetic testing is not indicated in relatives when the index patient does not have a definitive pathogenic mutation.[3][4][5][6][7][8][21] (Level of Evidence: B)” |
| ”2. Ongoing clinical screening is not indicated in genotype-negative relatives in families with HCM.[21][22][23][24] (Level of Evidence: B)” |
Genotype-Positive/Phenotype-Negative Patients (DO NOT EDIT) [2]
| Class I |
| “1. In individuals with pathogenic mutations who do not express the HCM phenotype, it is recommended to perform serial electrocardiogram (ECG), transthoracic echocardiogram (TTE), and clinical assessment at periodic intervals (12 to 18 months in children and adolescents and about every 5 years in adults), based on the patient’s age and change in clinical status.[25][26][27][28] (Level of Evidence: B)” |
Sources
2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy [29][2]
References
- ↑ Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P; et al. (2020). “2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines”. Circulation. 142 (25): e558–e631. doi:10.1161/CIR.0000000000000937. PMID 33215931 Check
|pmid=value (help). - ↑ 2.0 2.1 2.2 2.3 Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW (2011). “2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons”. Journal of the American College of Cardiology. 58 (25): e212–60. doi:10.1016/j.jacc.2011.06.011. PMID 22075469. Retrieved 2011-12-19. Unknown parameter
|month=ignored (help) - ↑ 3.0 3.1 3.2 3.3 Ho CY, Sweitzer NK, McDonough B, Maron BJ, Casey SA, Seidman JG, Seidman CE, Solomon SD (2002). “Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy”. Circulation. 105 (25): 2992–7. PMID 12081993. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ 4.0 4.1 4.2 Arad M, Maron BJ, Gorham JM, Johnson WH, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG (2005). “Glycogen storage diseases presenting as hypertrophic cardiomyopathy”. The New England Journal of Medicine. 352 (4): 362–72. doi:10.1056/NEJMoa033349. PMID 15673802. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ 5.0 5.1 5.2 Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG, Seidman CE (2008). “Shared genetic causes of cardiac hypertrophy in children and adults”. The New England Journal of Medicine. 358 (18): 1899–908. doi:10.1056/NEJMoa075463. PMC 2752150. PMID 18403758. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ 6.0 6.1 6.2 Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, Seidman JG, Seidman CE (1998). “Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy”. The New England Journal of Medicine. 338 (18): 1248–57. doi:10.1056/NEJM199804303381802. PMID 9562578. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ 7.0 7.1 7.2 Van Driest SL, Ackerman MJ, Ommen SR, Shakur R, Will ML, Nishimura RA, Tajik AJ, Gersh BJ (2002). “Prevalence and severity of “benign” mutations in the beta-myosin heavy chain, cardiac troponin T, and alpha-tropomyosin genes in hypertrophic cardiomyopathy”. Circulation. 106 (24): 3085–90. PMID 12473556. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ 8.0 8.1 8.2 Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ (2004). “Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy”. Journal of the American College of Cardiology. 44 (3): 602–10. doi:10.1016/j.jacc.2004.04.039. PMID 15358028. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Christiaans I, van Langen IM, Birnie E, Bonsel GJ, Wilde AA, Smets EM (2009). “Genetic counseling and cardiac care in predictively tested hypertrophic cardiomyopathy mutation carriers: the patients’ perspective”. American Journal of Medical Genetics. Part a. 149A (7): 1444–51. doi:10.1002/ajmg.a.32915. PMID 19533783. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Michie S, French D, Allanson A, Bobrow M, Marteau TM (1997). “Information recall in genetic counselling: a pilot study of its assessment”. Patient Education and Counseling. 32 (1–2): 93–100. PMID 9355576. Retrieved 2011-12-21.
- ↑ Michie S, Allanson A, Armstrong D, Weinman J, Bobrow M, Marteau TM (1998). “Objectives of genetic counselling: differing views of purchasers, providers and users”. Journal of Public Health Medicine. 20 (4): 404–8. PMID 9923946. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Offit K, Groeger E, Turner S, Wadsworth EA, Weiser MA (2004). “The “duty to warn” a patient’s family members about hereditary disease risks”. JAMA : the Journal of the American Medical Association. 292 (12): 1469–73. doi:10.1001/jama.292.12.1469. PMID 15383518. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Christiaans I, van Langen IM, Birnie E, Bonsel GJ, Wilde AA, Smets EM (2009). “Quality of life and psychological distress in hypertrophic cardiomyopathy mutation carriers: a cross-sectional cohort study”. American Journal of Medical Genetics. Part a. 149A (4): 602–12. doi:10.1002/ajmg.a.32710. PMID 19253387. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ 14.0 14.1 Fokstuen S, Lyle R, Munoz A, Gehrig C, Lerch R, Perrot A, Osterziel KJ, Geier C, Beghetti M, Mach F, Sztajzel J, Sigwart U, Antonarakis SE, Blouin JL (2008). “A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy”. Human Mutation. 29 (6): 879–85. doi:10.1002/humu.20749. PMID 18409188. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, Ommen SR, Theis JL, Vaubel RA, Re F, Armentano C, Poggesi C, Torricelli F, Cecchi F (2008). “Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy”. Mayo Clinic Proceedings. Mayo Clinic. 83 (6): 630–8. PMID 18533079. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Maron BJ, Niimura H, Casey SA, Soper MK, Wright GB, Seidman JG, Seidman CE (2001). “Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations”. Journal of the American College of Cardiology. 38 (2): 315–21. PMID 11499718. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Rosenzweig A, Watkins H, Hwang DS, Miri M, McKenna W, Traill TA, Seidman JG, Seidman CE (1991). “Preclinical diagnosis of familial hypertrophic cardiomyopathy by genetic analysis of blood lymphocytes”. The New England Journal of Medicine. 325 (25): 1753–60. doi:10.1056/NEJM199112193252501. PMID 1944483. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ (2006). “High incidence of later-onset fabry disease revealed by newborn screening”. American Journal of Human Genetics. 79 (1): 31–40. doi:10.1086/504601. PMC 1474133. PMID 16773563. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H (1997). “Sudden death due to troponin T mutations”. Journal of the American College of Cardiology. 29 (3): 549–55. PMID 9060892. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Woo A, Rakowski H, Liew JC, Zhao MS, Liew CC, Parker TG, Zeller M, Wigle ED, Sole MJ (2003). “Mutations of the beta myosin heavy chain gene in hypertrophic cardiomyopathy: critical functional sites determine prognosis”. Heart (British Cardiac Society). 89 (10): 1179–85. PMC 1767874. PMID 12975413. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ 21.0 21.1 Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE (2000). “Homozygous mutation in cardiac troponin T: implications for hypertrophic cardiomyopathy”. Circulation. 102 (16): 1950–5. PMID 11034944. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C (2005). “Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling”. Journal of Medical Genetics. 42 (10): e59. doi:10.1136/jmg.2005.033886. PMC 1735926. PMID 16199542. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ (2004). “Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy”. Journal of the American College of Cardiology. 44 (9): 1903–10. doi:10.1016/j.jacc.2004.07.045. PMID 15519027. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Jeschke B, Uhl K, Weist B, Schröder D, Meitinger T, Döhlemann C, Vosberg HP (1998). “A high risk phenotype of hypertrophic cardiomyopathy associated with a compound genotype of two mutated beta-myosin heavy chain genes”. Human Genetics. 102 (3): 299–304. PMID 9544842. Retrieved 2011-12-21. Unknown parameter
|month=ignored (help) - ↑ Christiaans I, Lekanne dit Deprez RH, van Langen IM, Wilde AA (2009). “Ventricular fibrillation in MYH7-related hypertrophic cardiomyopathy before onset of ventricular hypertrophy”. Heart Rhythm : the Official Journal of the Heart Rhythm Society. 6 (9): 1366–9. doi:10.1016/j.hrthm.2009.04.029. PMID 19539541. Retrieved 2011-12-22. Unknown parameter
|month=ignored (help) - ↑ Andersen PS, Havndrup O, Hougs L, Sørensen KM, Jensen M, Larsen LA, Hedley P, Thomsen AR, Moolman-Smook J, Christiansen M, Bundgaard H (2009). “Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives”. Human Mutation. 30 (3): 363–70. doi:10.1002/humu.20862. PMID 19035361. Retrieved 2011-12-22. Unknown parameter
|month=ignored (help) - ↑ Christiaans I, Birnie E, van Langen IM, van Spaendonck-Zwarts KY, van Tintelen JP, van den Berg MP, Atsma DE, Helderman-van den Enden AT, Pinto YM, Hermans-van Ast JF, Bonsel GJ, Wilde AA (2010). “The yield of risk stratification for sudden cardiac death in hypertrophic cardiomyopathy myosin-binding protein C gene mutation carriers: focus on predictive screening”. European Heart Journal. 31 (7): 842–8. doi:10.1093/eurheartj/ehp539. PMID 20019025. Retrieved 2011-12-22. Unknown parameter
|month=ignored (help) - ↑ Michels M, Soliman OI, Phefferkorn J, Hoedemaekers YM, Kofflard MJ, Dooijes D, Majoor-Krakauer D, Ten Cate FJ (2009). “Disease penetrance and risk stratification for sudden cardiac death in asymptomatic hypertrophic cardiomyopathy mutation carriers”. European Heart Journal. 30 (21): 2593–8. doi:10.1093/eurheartj/ehp306. PMID 19666645. Retrieved 2011-12-22. Unknown parameter
|month=ignored (help) - ↑ Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW (2011). “2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons”. Journal of the American College of Cardiology. 58 (25): 2703–38. doi:10.1016/j.jacc.2011.10.825. PMID 22075468. Retrieved 2011-12-19. Unknown parameter
|month=ignored (help)
Natural History, Complications and Prognosis
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Lakshmi Gopalakrishnan, M.B.B.S. [2] Soroush Seifirad, M.D.[3] Laith Adnan Allaham, M.D.[4]
Overview
The natural history of hypertrophic cardiomyopathy is quite variable. Signs and symptoms range from none, to atrial fibrillation, heart failure, embolic stroke and sudden cardiac death. Signs and symptoms are quite variable from individual to individual but are also quite variable within a given family (all of whom carry the same mutation). The disease is quite variable in the timing of its appearance and may appear anywhere from infancy to late in adult life. About 25% of HCM patients achieve normal longevity. The myosin-binding proteins C genetic variant carries a good prognosis. The presence of ventricular fibrillation/ ventricular tachycardia carries the poorest prognosis. The severity of the outflow gradient is also related to prognosis. Athletes should be screened for HOCM based upon a family history of sudden cardiac death and a murmur on physical examination. Electrocardiograms and echocardiograms are not cost-effective screening tools in this population with a low pre-test probability of disease.
Natural History, Complications and Prognosis
The natural history of hypertrophic cardiomyopathy is quite variable. Signs and symptoms range from none, to atrial fibrillation, heart failure, embolic stroke and sudden cardiac death.[1][2][3][4][5] Signs and symptoms are quite variable from individual to individual but are also quite variable within a given family (all of whom carry the same mutation). The disease is quite variable in the timing of its appearance and may appear anywhere from infancy to late in adult life. About 25% of HCM patients achieve normal longevity.[6][7][8][9]The myosin binding proteins C genetic variant carries a good prognosis. The presence of VT / VF carries the poorest prognosis. The severity of the outflow gradient is also related to prognosis. Athletes should be screened for HOCM based upon a family history of sudden cardiac death and a murmur on physical examination. Electrocardiograms and echocardiograms are not cost-effective screening tools in this population with a low pre-test probability of disease.
Time and Age-Dependent Appearance of Left Ventricular Hypertrophy
Left ventricular hypertrophy may be absent in childhood. It may then appear following the rapid growth of adolescence and may first appear at age 17 to 18.[10][11][12]
Sudden Cardiac Death
The incidence of sudden cardiac death (SCD) in patients with HCM is 2 to 4 percent per year in adults, and a 4 to 6 percent per year in children and adolescents.[13]
Among end stage patients with a left ventricular ejection fraction < 50%, the risk of SCD over 5 years may be as high as 47%. In this population, syncope has been identified as an independent correlate of sudden cardiac death (hazard ratio = 6.15; 95% confidence interval, 2.40-15.75; P < .001).[14]
A review of 78 patients with HCM who died suddenly or survived a cardiac arrest episode showed that 71 percent were younger than 30 years of age, 54 percent were without functional limitation, and 61 percent were performing a sedentary or minimal physical activity at the time of the cardiac arrest.
Predictors of Sudden Cardiac Death
There are few predictors of SCD in patients with HCM.
- Onset of symptoms in childhood. [15][16]
- A clinical history of spontaneous, sustained monomorphic ventricular tachycardia (VT) or sudden death in family members.
- History of impaired consciousness
- Syncope
- Atrial arrhythmias
- Development of systolic dysfunction
- Non-sustained ventricular tachycardia (NSVT) in patients with symptoms
- Left ventricular wall thickness >30 mm. A recent report of 480 patients showed that left ventricular wall thickness was useful in identifying patients at high risk for sudden cardiac death. However, sudden cardiac death can occur in children and adolescents in the absence of left ventricular hypertrophy as well.
2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines[17]
Recommendations for SCD Risk Assessment Referenced studies that support the recommendations are summarized in Online Data Supplement
| Class I |
| 1. In patients with HCM, a comprehensive, systematic noninvasive SCD risk assessment at initial evaluation and every 1 to 2 years thereafter is recommended and should include evaluation of these risk factors1–25 (Figure 1 and Figure 3, Table 7):(Level of Evidence: B-NR)
a. Personal history of cardiac arrest or sustained ventricular arrhythmias b. Personal history of syncope suspected by clinical history to be arrhythmic c. Family history in close relative of premature HCM-related sudden death, cardiac arrest, or sustained ventricular arrhythmias d. Maximal LV wall thickness, EF, LV apical aneurysm e. NSVT episodes on continuous ambulatory electrocardiographic monitoring |
| Class IIa |
| 3. For patients who are ≥ 16 years of age with HCM, it is reasonable to obtain echocardiography-derived left atrial diameter and maximal LVOT gradient to aid in calculating an estimated 5-year sudden death risk that may be useful during shared decision-making for ICD placement(Level of Evidence: B-NR) |
ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death – Hypertrophic Cardiomyopathy (DO NOT EDIT) [18]
| Class I |
| “1. ICD therapy should be used for treatment in patients with HCM who have sustained VT and/or VF and who are receiving chronic optimal medical therapy and who have reasonable expectation of survival with a good functional status for more than 1 y. (Level of Evidence: B)” |
| Class IIa |
| “1. ICD implantation can be effective for primary prophylaxis against SCD in patients with HCM who have 1 or more major risk factor (see Table 7) for SCD and who are receiving chronic optimal medical therapy and in patients who have reasonable expectation of survival with a good functional status for more than 1 y. (Level of Evidence: C)” |
| “2. Amiodarone therapy can be effective for treatment in patients with HCM with a history of sustained VT and/or VF when an ICD is not feasible. (Level of Evidence: C)” |
| Class IIb |
| “1. EP testing may be considered for risk assessment for SCD in patients with HCM. (Level of Evidence: C)” |
| “2. Amiodarone may be considered for primary prophylaxis against SCD in patients with HCM who have 1 or more major risk factor for SCD (see Table 7) if ICD implantation is not feasible. (Level of Evidence: C)” |
Prognosis in Survivors of Sudden Cardiac Death
Survivors of SCD have a poor prognosis. Event free survival at 1, 5, and 10 years was 83, 65, and 53 percent respectively.
2011 ACCF/AHA Guideline Recommendations: SCD Risk Stratification
| Class I |
| “1. All patients with HCM should undergo comprehensive SCD risk stratification at initial evaluation to determine the presence of the following: ” |
| “a. A personal history for ventricular fibrillation, sustained VT, or SCD events, including appropriate ICD therapy for ventricular tachyarrhythmias.† (Level of Evidence: B)” |
| “b. A family history for SCD events, including appropriate ICD therapy for ventricular tachyarrhythmias.† (Level of Evidence: B)” |
| “c. Unexplained syncope. (Level of Evidence: B)” |
| “d. Documented NSVT defined as 3 or more beats at greater than or equal to 120 bpm on ambulatory (Holter) ECG. (Level of Evidence: B) ” |
| “e. Maximal LV wall thickness greater than or equal to 30 mm. (Level of Evidence: B) ” |
| Class III (Harm) |
| “1. Invasive electrophysiologic testing as routine SCD risk stratification for patients with HCM should not be performed. (Level of Evidence: C) ” |
| Class IIa |
| “1. It is reasonable to assess blood pressure response during exercise as part of SCD risk stratification in patients with HCM.(89,127,390) (Level of Evidence: B) ” |
| “2. SCD risk stratification is reasonable on a periodic basis (every 12 to 24 months) for patients with HCM who have not undergone ICD implantation but would otherwise be eligible in the event that risk factors are identified (12 to 24 months).(Level of Evidence: C) ” |
| Class IIb |
| “1. The usefulness of the following potential SCD risk modifiers is unclear but might be considered in selected patients with HCM for whom risk remains borderline after documentation of conventional risk factors:” |
| “a. CMR imaging with LGE.(184,188) (Level of Evidence: C)” |
| “b. Double and compound mutations (i.e., >1). (Level of Evidence: C) ” |
| “c. Marked LVOT obstruction.(45,127,143,390) (Level of Evidence: B) ” |
Guideline Resources
2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy [19][20]
References
- ↑ Maron BJ. Hypertrophic cardiomyopathy. Lancet 1997;350:127–33.
- ↑ Maron BJ. Hypertrophic cardiomyopathy. A systematic review. JAMA 2002;287:1308–20.
- ↑ Maki S, Ikeda H, Muro A et al. Predictors of sudden cardiac death in hypertrophic cardiomyopathy. Am J Cardiol 1998;82:774–8.
- ↑ Maron BJ, Casey SA, Poliac LC, Gohman TE, Almquist AK, Aeppli DM. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA 1999;281:650–5.
- ↑ Maron BJ, Olivotto I, Bellone P et al. Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002;39:301–7.
- ↑ Maron BJ. Hypertrophic cardiomyopathy. A systematic review. JAMA 2002;287:1308–20.
- ↑ Maron BJ, Casey SA, Poliac LC, Gohman TE, Almquist AK, Aeppli DM. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA 1999;281:650–5.
- ↑ Fay WP, Taliercio CP, Ilstrup DM, Tajik AJ, Gersh BJ. Natural history of hypertrophic cardiomyopathy in the elderly. J Am Coll Cardiol 1990;16:821–6.
- ↑ Takagi E, Yamakado T, Nakano T. Prognosis of completely asymptomatic adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999;33:206–11.
- ↑ Hagege AA, Dubourg O, Desnos M et al. Familial hypertrophic cardiomyopathy. Cardiac ultrasonic abnormalities in genetically affected subjects without echocardiographic evidence of left ventricular hypertrophy. Eur Heart J 1998;19:490–9.
- ↑ Maron BJ, Spirito P, Wesley Y, Arce J. Development and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med 1986;315:610–4.
- ↑ Spirito P, Maron BJ. Absence of progression of left ventricular hypertrophy in adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1987;9:1013–7.
- ↑ Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A; et al. (2000). “Sudden death in hypertrophic cardiomyopathy: identification of high risk patients”. J Am Coll Cardiol. 36 (7): 2212–8. PMID 11127463.
- ↑ Kawarai H, Kajimoto K, Minami Y, Hagiwara N, Kasanuki H (2011). “Risk of sudden death in end-stage hypertrophic cardiomyopathy”. J Card Fail. 17 (6): 459–64. doi:10.1016/j.cardfail.2011.01.015. PMID 21624733.
- ↑ Maron BJ, Tajik AJ, Ruttenberg HD et al. Hypertrophic cardiomyopathy in infants. Clinical features and natural history. Circulation 1982; 65:7–17
- ↑ Skinner JR, Manzoor A, Hayes AM, Joffe HS, Martin RP. A regional study of presentation and outcome of hypertrophic cardiomyopathy in infants. Heart 1997;77:229–33.
- ↑ Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P; et al. (2020). “2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines”. Circulation. 142 (25): e558–e631. doi:10.1161/CIR.0000000000000937. PMID 33215931 Check
|pmid=value (help). - ↑ Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M; et al. (2006). “ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society”. Circulation. 114 (10): e385–484. doi:10.1161/CIRCULATIONAHA.106.178233. PMID 16935995.
- ↑ 19.0 19.1 Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW (2011). “2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons”. Journal of the American College of Cardiology. 58 (25): 2703–38. doi:10.1016/j.jacc.2011.10.825. PMID 22075468. Retrieved 2011-12-19. Unknown parameter
|month=ignored (help) - ↑ 20.0 20.1 Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW (2011). “2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons”. Journal of the American College of Cardiology. 58 (25): e212–60. doi:10.1016/j.jacc.2011.06.011. PMID 22075469. Retrieved 2011-12-19. Unknown parameter
|month=ignored (help)
Diagnosis
Diagnosis
Diagnostic Study of Choice| History and Symptoms | Physical examination | Laboratory findings | Electrocardiogram | X Ray | Echocardiography and Ultrasound | CT Scan | MRI |Other Imaging Findings | Other Diagnostic Studies.
Treatment
Treatment
- Medical therapy, Interventions, Surgery, Primary Prevention, Secondary Prevention, Cost Effectiveness of Therapy, Future or Investigational Therapy
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH