
Multiple endocrine neoplasia type 2
For patient information, click here.
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Synonyms and keywords: Familial endocrine adenomatosis type 2; Familial endocrine adenomatosis type 2a; Familial endocrine adenomatosis type 2b; Familial chromaffinomatosis type 2; Familial medullary thyroid carcinoma; Multiple endocrine adenomatosis type 2; Multiple endocrine adenomatosis type 2a; Multiple endocrine adenomatosis type 2b; Multiple endocrine neoplasia syndrome type 2; Multiple endocrine neoplasia syndrome type 2a; Multiple endocrine neoplasia syndrome type 2b; MEN type 2; MEN type 2a; MEN type 2b; MEN 2 syndrome; MEN type II; MEN type IIa; MEN type IIb; MEA type 2; MEA type 2a; MEA type 2b; Multiple endocrine neoplasms type 2; Multiple endocrine neoplasms type 2a; Multiple endocrine neoplasms type 2b; Multiple neuroma syndrome type 2; Multiple neuroma syndrome type 2a; Multiple neuroma syndrome type 2b; PTC syndrome; Sipple’s syndrome; Pheochromocytoma and amyloid producing medullary thyroid carcinoma; Wagenmann-Froboese syndrome
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Multiple endocrine neoplasia type 2 is part of a group of disorders that affect the endocrine system through development of neoplastic lesions in the thyroid, the parathyroid gland, and the adrenal gland. Multiple endocrine neoplasia type 2 was first described by Dr. John H. Sipple, an American physician, in 1961 by reporting a case of a patient with pheochromocytoma, medullary thyroid carcinoma and parathyroid adenoma. Development of multiple endocrine neoplasia type 2 is the result of genetic mutations. RET gene is involved in the pathogenesis of multiple endocrine neoplasia type 2. Multiple endocrine neoplasia type 2A is associated with medullary thyroid carcinoma, pheochromocytoma, and hyperparathyroidism. Multiple endocrine neoplasia type 2B is associated with medullary thyroid carcinoma, pheochromocytoma, marfanoid habitus, and mucosal and digestive neurofibromatosis. Surgery is the mainstay of treatment for multiple endocrine neoplasia type 2.
Historical Perspective
Multiple endocrine neoplasia type 2 was first described by Dr. John H. Sipple, an American physician, in 1961 by reporting a case of a patient with pheochromocytoma, medullary thyroid carcinoma, and parathyroid adenoma.
Classification
Multiple endocrine neoplasia type 2 may be classified according to the types of tumor involved into 2 subtypes: multiple endocrine neoplasia type 2A and multiple endocrine neoplasia type 2B.
Pathophysiology
Development of multiple endocrine neoplasia type 2 is the result of genetic mutations. The RET gene is involved in the pathogenesis of multiple endocrine neoplasia. Multiple endocrine neoplasia type 2A is associated with medullary thyroid carcinoma, pheochromocytoma, and hyperparathyroidism. Multiple endocrine neoplasia type 2B is associated with medullary thyroid carcinoma, pheochromocytoma, marfanoid habitus, and mucosal and digestive neurofibromatosis.
Causes
Multiple endocrine neoplasia type 2 is caused by a mutation in the RET gene.
Differentiating Multiple endocrine neoplasia type 2 from Other Diseases
Multiple endocrine neoplasia type 2 must be differentiated from other hereditary tumors such as medullary thyroid carcinoma, C-cell hyperplasia pheochromocytoma, von Hippel Lindau syndrome, hereditary paraganglioma–pheochromocytoma, polycythemia and paraganglioma/pheochromocytoma syndrome, neurofibromatosis type 1, and multiple endocrine neoplasia type 1 (MEN 1).
Epidemiology and Demographics
The prevalence of multiple endocrine neoplasia type 2 is approximately 2.5 per 100,000 individuals worldwide. Multiple endocrine neoplasia type 2B manifest in early infancy while multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma manifest in adulthood.
Risk Factors
Family history is the most common risk factor in the development of multiple endocrine neoplasia type 2.
Screening
According to the American Society of Clinical Oncology, screening for multiple endocrine neoplasia type 2 by RET gene testing is recommended for children with increased risk of multiple endocrine neoplasia type 2.
Natural history, Complications and Prognosis
Multiple endocrine neoplasia type 2 has a variable natural history. Life threatening complications of multiple endocrine neoplasia type 2 include malignant hypertension, megacolon, and metastasis. Prognosis of multiple endocrine neoplasia type 2 is mainly related to the stage-dependant prognosis of medullary thyroid cancer.
Diagnosis
Diagnostic Criteria
According to the American Society of Clinical Oncology, diagnosis of multiple endocrine neoplasia type 2 requires existence of two or more specific endocrine tumors (medullary thyroid carcinoma, pheochromocytoma, or parathyroid hyperplasia). Multiple endocrine neoplasia type 2 is also diagnosed with four or more cases of medullary thyroid carcinoma without pheochromocytoma and parathyroid adenoma or if there is early onset of medullary thyroid carcinoma, mucosal neuromas of lips and tongue, disctinctive facial features with enlarged lips and marfanoid body habitus.
History and Symptoms
Symptoms of multiple endocrine neoplasia type 2 include headache, hoarseness of voice, palpitations, anxiety, pallor and weight loss.
Physical Examination
Patients with multiple endocrine neoplasia type 2 usually appear tall, thin and have disproportionately elongated extremities. Physical examination of patients with multiple endocrine neoplasia type 2 is usually remarkable for episodic hypertension, thyromegaly, and anxiety.
Laboratory Findings
Laboratory findings consistent with the diagnosis of multiple endocrine neoplasia type 2 include hypercalcemia, hypophosphatemia, elevated parathyroid hormone, and elevated norepinephrine.
Electrocardiogram
There are no specific electrocardiogram findings associated with multiple endocrine neoplasia type 2. Electrocardiogram findings in multiple endocrine neoplasia type 2 may vary depending on the underlying disease.
CT
Neck CT scan may be helpful in the diagnosis of multiple endocrine neoplasia type 2. Findings on CT scan suggestive of multiple endocrine neoplasia type 2 include irregular dense calcific foci within thyroid.
MRI
MRI scan may be helpful in the diagnosis of multiple endocrine neoplasia type 2. Findings on MRI scan suggestive of multiple endocrine neoplasia type 2 include intermediate to low signal at T1 and hyperintense signal at T2 suggesting parathyroid hyperplasia.
Echocardiography or Ultrasound
Ultrasound scan may be helpful in the diagnosis of multiple endocrine neoplasia type 2. Findings on ultrasound scan suggestive of multiple endocrine neoplasia type 2 include punctate high echogenic foci resembling calcification within the thyroid gland, solid to mixed cystic masses on adrenal gland, and homogeneously hypoechoic parathyroid gland.
Other Imaging Findings
Other imaging studies for multiple endocrine neoplasia type 2 include fluoro-di-glucose-PET, [18F]-fluorodopamine ([18F]DA) PET, and 99mTc-sestamibi scintigraphy.
Other Diagnostic Studies
Other diagnostic studies for multiple endocrine neoplasia type 2 include molecular genetic testing which include sequence analysis of select exons of mainly 10,11, 13-16 and sequence analysis of RET gene.
Treatment
Medical Therapy
The mainstay of multiple endocrine neoplasia type 2 is surgery. Medical therapies for multiple endocrine neoplasia type 2 include vandetanib, external beam radiation therapy analogues, and intensity modulated radiation therapy.
Surgery
Surgery is the mainstay of treatment for multiple endocrine neoplasia type 2.
Primary Prevention
There are no primary preventive measures for multiple endocrine neoplasia type 2.
Secondary Prevention
According to the American Society of Clinical Oncology, surveillance for multiple endocrine neoplasia type 2 by annual measurement of serum calcitonin, serum calcium, serum parathyroid hormone, and catecholamines is recommended post-surgically to monitor for recurrence and complications for multiple endocrine neoplasia type 2.
Cost-effectiveness of the Therapy
Cost-effectiveness of therapy of multiple endocrine neoplasia type 2 include targeted therapy, prophylactic thyroidectomy and restricted molecular screening.
Future or Investigational therapies
Future or investigational therapies of multiple endocrine neoplasia type 2 include treatment with axitinib, gefitinib, imatinib, motesanib, sorafenib, sunitinib, vandetanib and XL184.
Reference
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Sogand Goudarzi, MD [2] , Ammu Susheela, M.D. [3]
Overview
Multiple endocrine neoplasia type 2 was first described by Dr. John H. Sipple, an American physician, in 1961 by reporting a case of a patient with pheochromocytoma, medullary thyroid carcinoma, and parathyroid adenoma.
Historical Perspective
The historical background of multiple endocrine neoplasia type 2 is given in the table below:[1][2][3][4][5]
| Years | Scientist | Contribution |
|---|---|---|
| 1954 | Wermer | Reported that syndrome was transmitted as a dominant trait |
| 1959 | Hazard | Described medullary (solid) thyroid carcinoma |
| 1961 | Sipple | Described a case of a patient with pheochromocytoma, medullary thyroid carcinoma, and parathyroid adenoma. It was based on a case he saw when he was in 3rd year medical residency about a person with intracranial bleed and fluctuating blood pressure. His autopsy showed parathyroid tumor, thyroid tumor, and bilateral adrenal pheochromocytomas. |
| 1965 | Schimke and Hartmann | Described a syndrome of medullary thyroid carcinoma with abundant amyloid stroma and pheochromocytoma |
| 1966 | Williams | Reported a case of a patient with combination of mucosal neuromas, pheochromocytoma, and medullary thyroid carcinoma |
| 1968 | Steiner | Introduced the term “multiple endocrine neoplasia” (MEN) to describe disorders featuring combinations of endocrine tumors and proposed the terms ‘Wermer syndrome‘ for multiple endocrine neoplasia type 1 and ‘Sipple syndrome‘ for multiple endocrine neoplasia type 2 |
| Meyer and Abdel-Bari | Suggested that medullary carcinoma produces thyrocalcitonin from parafollicular cells | |
| 1970 | Kaplan | Suggested that adrenal medulla produces a calcitonin like material |
| 1974 | Sizemore | Showed that the multiple endocrine neoplasia type 2 category included two groups of patients with medullary thyroid cancer and pheochromocytoma: one with parathyroid disease and a normal appearance (multiple endocrine neoplasia type 2A) and the other without parathyroid disease but with mucosal neuromas and mesodermal abnormalities (multiple endocrine neoplasia type 2B) |
| 1978 | Hamilton | Reported a case of Zollinger-Ellison syndrome in multiple endocrine hyperplasia type 2 |
| Cameron | Suggested that medullary carcinoma produces thyrocalcitonin from parafollicular cells | |
| 1980 | Le Marec | Described a case of congential megacolon in Sipple syndrome |
| 1989 | Sobol | Proposed that restriction fragment length polymerase is useful in predicting the carrier state of multiple endocrine neoplasia syndrome |
| 1993 | RET germline mutations were recognized as the causative molecular alterations in multiple endocrine neoplasia type 2 syndromes | |
| 1998 | MEN1 gene was cloned | |
|
2000-2001 |
Huang,Koch |
Introduced the 2 hit mechanism for multiple endocrine neoplasia type 2 associated tumors and also described the mechanism of involved in trisomy 10 |
References
- ↑ Schimke RN, Hartmann WH (1965). “Familial amyloid-producing medullary thyroid carcinoma and pheochromocytoma. A distinct genetic entity”. Ann Intern Med. 63 (6): 1027–39. PMID 5844561.
- ↑ Kaplan EL, Arnaud CD, Hill BJ, Peskin GW (1970). “Adrenal medullary calcitonin-like factor: a key to multiple endocrine neoplasia, type 2?”. Surgery. 68 (1): 146–9. PMID 10483461.
- ↑ Cameron D, Spiro HM, Landsberg L (1978). “Zollinger-Ellison syndrome with multiple endocrine adenomatosis type II”. N Engl J Med. 299 (3): 152–3. doi:10.1056/NEJM197807202990315. PMID 26873.
- ↑ Le Marec B, Roussey M, Cornec A, Calmettes C, Kerisit J, Allanic H (1981). “[Thyroid cancer with amyloid stroma, Sipple’s syndrome, congenital megacolon with plexus hyperplasia: one and the same dominant autosomal disease with complete penetrance]”. J Genet Hum. 28 (5): 169–74. PMID 7276917.
- ↑ Guru SC, Manickam P, Crabtree JS, Olufemi SE, Agarwal SK, Debelenko LV. Identification and characterization of the multiple endocrine neoplasia type 1 (MEN1) gene. J Intern Med 243(6) 433-9
Classification
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2] Sogand Goudarzi, MD [3]
Overview
Multiple endocrine neoplasia type 2 may be classified according to the types of tumor involved into 2 subtypes: multiple endocrine neoplasia type 2A and multiple endocrine neoplasia type 2B.
Classification
Multiple endocrine neoplasia type 2 (MEN2) can be further divided into three subtypes: type 2A, type 2B, and familial medullary thyroid carcinoma (FMTC).[1][2]
- These subtypes differ in their characteristic signs and symptoms and risk of specific tumors, but are relatively consistent within any one family.
- Multiple endocrine neoplasia type 2A is characterized by the presence of medullary thyroid carcinoma, pheochromocytoma, and parathyroid hyperplasia or tumor.
- Multiple endocrine neoplasia type 2B is characterized by the presence of pheochromocytoma, mucocutaneous neuroma, and medullary thyroid cancer.
- The following flowchart depicts the classification of multiple endocrine neoplasia type 2.
| Multiple endocrine neoplasia type 2 | |||||||||||||||||||||||||||||||||||||||||
| Multiple endocrine neoplasia type 2A | Multiple endocrine neoplasia type 2B | ||||||||||||||||||||||||||||||||||||||||
| Multiple endocrine neoplasia type 2A classical | Multiple endocrine neoplasia type 2A with cutaneous lichen amyloidosis | Multiple endocrine neoplasia type 2A with Hirschsprung disease | Familial medullary thyroid carcinoma without pheochromocytoma or parathyroid hyperplasia | Medullary thyroid cancer, pheochromocytoma,marfanoid habitus, and mucosal neuromas or intestinal ganglioneuromas | |||||||||||||||||||||||||||||||||||||
References
- ↑ Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF; et al. (2015). “Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma”. Thyroid. 25 (6): 567–610. doi:10.1089/thy.2014.0335. PMC 4490627. PMID 25810047.
- ↑ Hansford, J. R (2000). “Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis”. Journal of Medical Genetics. 37 (11): 817–827. doi:10.1136/jmg.37.11.817. ISSN 1468-6244.
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [2]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [3]
Overview
The progression to multiple endocrine neoplasia type 2 usually involves the genetic mutations. The pathogenesis of multiple endocrine neoplasia type 2 involves a mutation of the RET gene.
Pathogenesis
The common feature among the three subtypes of multiple endocrine neoplasia type 2 is a high propensity to develop medullary thyroid carcinoma.
Multiple Endocrine Neoplasia type 2A
- Multiple endocrine neoplasia type 2 (MEN2) generally occur in endocrine organs (e.g. thyroid, parathyroid, and adrenals), but may also occur in endocrine tissues of organs not classically thought of as endocrine.[1]
- Although many different types of hormone-producing tumors are associated with multiple endocrine neoplasia type 2, the most common manifestation is a form of thyroid cancer called medullary thyroid carcinoma. This tumor secretes an inactive hormone called calcitonin. [2]
- Many people with this disorder also develop pheochromocytoma, which is a tumor of the adrenal glands (located above each kidney) that can cause dangerously high blood pressure. In addition, overactivity of the parathyroid gland (hyperparathyroidism) occurs in some cases of multiple endocrine neoplasia type 2. Hyperparathyroidism disrupts the normal balance of calcium in the blood, which can lead to kidney stones, thinning of bones, weakness, and fatigue.[3]
- In multiple endocrine neoplasia type 2, primary hyperparathyroidism occurs in only 10–30% and is usually diagnosed after the third decade of life. It can occur in children but this is rare. It may be the sole clinical manifestation of this syndrome but this is unusual.[4]
- Multiple endocrine neoplasia type 2 associates medullary thyroid carcinoma with pheochromocytoma in about 20–50% of cases and with primary hyperparathyroidism in 5–20% of cases.[5]
- Other components of the disease are absent in familial isolated medullary thyroid carcinoma.[6]
Multiple Endocrine Neoplasia type 2B
- Multiple endocrine neoplasia type 2B is associated with medullary thyroid carcinoma and pheochromocytoma as well as with marfanoid habitus and with mucosal and digestive neurofibromatosis.
| Subtype | Medullary Thyroid Carcinoma | Pheochromocytoma | Parathyroid Disease |
|---|---|---|---|
| Multiple endocrine neoplasia type 2A | 95% | 50% | 20% to 30% |
| Multiple endocrine neoplasia type 2B | 100% | 50% | Uncommon |
| Familial medullary thyroid carcinoma | 100% | 0% | 0% |
Genetics

- Most cases of multiple endocrine neoplasia type 2 are inherited in an autosomal dominant pattern, which means affected people may have affected siblings and relatives in successive generations (such as parents and children). An affected person usually has one parent with the condition. Some cases, however, result from new mutations in the RET proto-oncogene. These cases occur in people with no history of the disorder in their family.

- Germline mutations are responsible for sporadic multiple endocrine neoplasia type 2, while mutations in the cysteine residues in the exons of the RET protein product are common in familial multiple endocrine neoplasia type 2.[7]
- The majority of multiple endocrine neoplasia type 2 patients, derive from a variation in the RET proto-oncogene, and are specific for cells of neural crest origin.[8]
- The RET protooncogene is a 21-exon gene and encodes for a tyrosine kinase transmembrane receptor located on chromosome 10q11.2.[9]
- Four different ligands have so far been recognized: The glial cell line-derived neutrophilic factor (GDNF), neurturin (NTN), persepin (PNS) and artemin (ART). The interaction is mediated by a ligand-specific coreceptor (e.g., the GFRα-1 is the co-receptor for the GDNF).[10]
- The receptor is composed of an extracellular domain (EC), with a distal cadherin-like region and a juxtamembrane cysteine-rich region, a transmembrane domain (TM) and an intracellular domain with tyrosine-kinase activity (TK).[11]
The table below summarizes specific RET codons and their functions.[12][13]
| Mutation location | RET codons | Function of wild type codon | Mutated effects | Phenotype |
|---|---|---|---|---|
| Extracellular cysteine rich location |
|
Helps to form teritiary structure with the help of disulfide bonds | Alteration in protein folding and maturation | Multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma (FMTC) |
| c634 | Formation of intramolecular disulfide bonds | Ligand independant dimerization of receptor molecules | Multiple endocrine neoplasia type 2A | |
| Intracellular tyrosine kinase domain |
|
Terminal lobe of RET kinase | Affects ATP binding and interlobe flexibility | Multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma (FMTC) |
|
Close proximity with ATP binding site | Alters interactions within the region | Familial medullary thyroid carcinoma (FMTC) | |
|
Gatekeeper residue that regulates access to ATP binding site | Alters interactions within the region | Familial medullary thyroid carcinoma (FMTC) | |
|
C terminal lobe of kinase | Alters activation of loop conformation | Multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma (FMTC) | |
|
Situated next to activated loop | Local conformational change | Multiple endocrine neoplasia type 2B | |
|
Substrate binding pocket of the kinase | Alters protein conformation | Multiple endocrine neoplasia type 2B |
- Multiple endocrine neoplasia type 2 generally results from a gain-of-function variant of a RET gene. Other diseases, such as Hirschsprung disease, result from loss-of-function variants.[14]
- Multiple endocrine neoplasia type 2 is transmitted in an autosomal dominant. Nevertheless, it can result from spontaneous new mutations in the RET gene with no family history of the disorder, as reported in some cases. For instance, among multiple endocrine neoplasia type 2B, spontaneous new mutations were observed in about 50% of the total number of cases.
- Activating germline point mutations of the RET proto-oncogene are causative events in multiple endocrine neoplasia type 2A, multiple endocrine neoplasia type 2B, and familial medullary thyroid carcinoma (FMTC). RET mutations have been found to be widely distributed not only among the 5 cysteine codons 609, 611, 618, 620, and 634 but also in other noncysteine codons, such as codon 804 in exon 14, codon 883 in exon 15, and others.[15]
- The following figure depicts the structure and mutation of RET receptor.[12]

RET Activation
- Dimerization of RET proto-oncogene mediated through formation of multicomponent complex. RET proto-oncogene is activated by binding both a soluble ligand and a non signaling extracellular co-receptor. RET activation leads to phosphorylation of multiple intracellular tyrosine kinase.

Associated Conditions
- Some of the diseases specific to the genes of multiple endocrine neoplasia type 2 are as follows.[12]
| Subtype | Associated diseases |
|---|---|
| Multiple endocrine neoplasia type 2A | Cutaneous lichen amyloidosis, Hirschsprung disease |
| Multiple endocrine neoplasia type 2B | Ganglioneuromatosis, marfanoid habitus |
| Familial medullary thyroid carcinoma (FMTC) | Rare diseases |
Gross Pathology
-
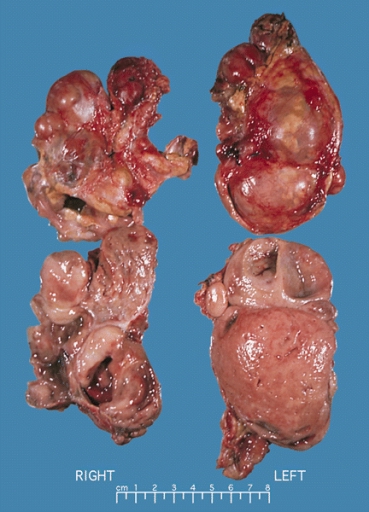 ADRENAL GLAND: BILATERAL PHEOCHROMOCYTOMA Cross section of bilateral pheochromocytomas from a 30-year-old man with MEN syndrome type IIa. The right adrenal tumor weighed 168 g and the left 220 g. Note the distinct multinodular, multicentric pattern of growth on both sides Source: Wikimedia Commons
ADRENAL GLAND: BILATERAL PHEOCHROMOCYTOMA Cross section of bilateral pheochromocytomas from a 30-year-old man with MEN syndrome type IIa. The right adrenal tumor weighed 168 g and the left 220 g. Note the distinct multinodular, multicentric pattern of growth on both sides Source: Wikimedia Commons -
![Pheochromocytoma, Image courtesy of Dr Frank Gaillard[16]](https://www.wikidoc.org/images/3/38/Pheochromocytoma_03.jpeg) Pheochromocytoma, Image courtesy of Dr Frank Gaillard[16]
Pheochromocytoma, Image courtesy of Dr Frank Gaillard[16] -
![Pheochromocytoma, Image courtesy of Dr Frank Gaillard[16]](https://www.wikidoc.org/images/6/6a/Pheochromocytoma_04.JPG) Pheochromocytoma, Image courtesy of Dr Frank Gaillard[16]
Pheochromocytoma, Image courtesy of Dr Frank Gaillard[16]

![Pheochromocytoma, Image courtesy of Dr Frank Gaillard[16]](https://www.wikidoc.org/index.php/File%3APheochromocytoma_03.jpeg)
![Pheochromocytoma, Image courtesy of Dr Frank Gaillard[16]](https://www.wikidoc.org/index.php/File%3APheochromocytoma_04.JPG)
Microscopic Pathology
Medullary Carcinoma of Thyroid
- Nests of C cells invading the basement membrane and infiltrating thyroid follicles
-
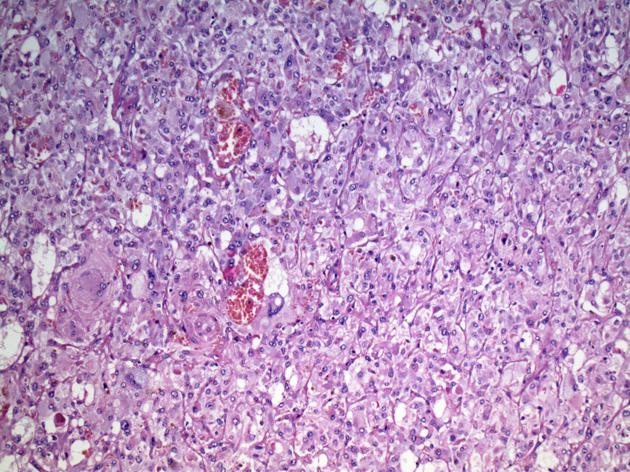 Pheochromocytoma, Case courtesy of Dr Andrew Ryan, Radiopaedia.org From the case <a href=”http://radiopaedia.org/cases/22683″>rID: 22683
Pheochromocytoma, Case courtesy of Dr Andrew Ryan, Radiopaedia.org From the case <a href=”http://radiopaedia.org/cases/22683″>rID: 22683

Histopathological Video
Video
{{#ev:youtube|P2uPUbDPbuI}}
{{#ev:youtube|7yjxG3KmX98}}
{{#ev:youtube|crwGfnWKEZ8}}
References
- ↑ Moline J, Eng C. (2011). “Multiple endocrine neoplasia type 2: an overview”. Genet Med. 9 (13): 755–64. doi:10.1097/GIM.0b013e318216cc6d. PMID 21552134.
- ↑ Cote, Gilbert J.; Grubbs, Elizabeth G.; Hofmann, Marie-Claude (2015). “Thyroid C-Cell Biology and Oncogenic Transformation”. 204: 1–39. doi:10.1007/978-3-319-22542-5_1. ISSN 0080-0015.
- ↑ Kantorovich, Vitaly; Pacak, Karel (2010). “Pheochromocytoma and Paraganglioma”. 182: 343–373. doi:10.1016/S0079-6123(10)82015-1. ISSN 0079-6123.
- ↑ Wells, Samuel A.; Pacini, Furio; Robinson, Bruce G.; Santoro, Massimo (2013). “Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma: An Update”. The Journal of Clinical Endocrinology & Metabolism. 98 (8): 3149–3164. doi:10.1210/jc.2013-1204. ISSN 0021-972X.
- ↑ Almeida, Madson Q.; Stratakis, Constantine A. (2010). “Solid tumors associated with multiple endocrine neoplasias”. Cancer Genetics and Cytogenetics. 203 (1): 30–36. doi:10.1016/j.cancergencyto.2010.09.006. ISSN 0165-4608.
- ↑ Niccoli-Sire, P.; Conte-Devolx, B. (2007). “Néoplasies endocriniennes multiples de type 2”. Annales d’Endocrinologie. 68 (5): 317–324. doi:10.1016/j.ando.2007.04.005. ISSN 0003-4266.
- ↑ Wells, Samuel A.; Pacini, Furio; Robinson, Bruce G.; Santoro, Massimo (2013). “Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma: An Update”. The Journal of Clinical Endocrinology & Metabolism. 98 (8): 3149–3164. doi:10.1210/jc.2013-1204. ISSN 0021-972X.
- ↑ Martucciello, Giuseppe; Lerone, Margherita; Bricco, Lara; Tonini, Gian; Lombardi, Laura; Del Rossi, Carmine G; Bernasconi, Sergio (2012). “Multiple endocrine neoplasias type 2B and RET proto-oncogene”. Italian Journal of Pediatrics. 38 (1): 9. doi:10.1186/1824-7288-38-9. ISSN 1824-7288.
- ↑ C. Romei, E. Pardi, F. Cetani, and R. Elisei, “Genetic and Clinical Features of Multiple Endocrine Neoplasia Types 1 and 2,” Journal of Oncology, vol. 2012, Article ID 705036, 15 pages, 2012. doi:10.1155/2012/705036
- ↑ Schmutzler, B.S.; Roy, S.; Hingtgen, C.M. (2009). “Glial cell line–derived neurotrophic factor family ligands enhance capsaicin-stimulated release of calcitonin gene-related peptide from sensory neurons”. Neuroscience. 161 (1): 148–156. doi:10.1016/j.neuroscience.2009.03.006. ISSN 0306-4522.
- ↑ De Falco, Valentina; Carlomagno, Francesca; Li, Hong-yu; Santoro, Massimo (2017). “The molecular basis for RET tyrosine-kinase inhibitors in thyroid cancer”. Best Practice & Research Clinical Endocrinology & Metabolism. 31 (3): 307–318. doi:10.1016/j.beem.2017.04.013. ISSN 1521-690X.
- ↑ 12.0 12.1 12.2 Wagner SM, Zhu S, Nicolescu AC, Mulligan LM (2012). “Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2”. Clinics (Sao Paulo). 67 Suppl 1: 77–84. PMC 3328826. PMID 22584710.
- ↑ Wells, Samuel A.; Pacini, Furio; Robinson, Bruce G.; Santoro, Massimo (2013). “Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma: An Update”. The Journal of Clinical Endocrinology & Metabolism. 98 (8): 3149–3164. doi:10.1210/jc.2013-1204. ISSN 0021-972X.
- ↑ Schmutzler, B.S.; Roy, S.; Hingtgen, C.M. (2009). “Glial cell line–derived neurotrophic factor family ligands enhance capsaicin-stimulated release of calcitonin gene-related peptide from sensory neurons”. Neuroscience. 161 (1): 148–156. doi:10.1016/j.neuroscience.2009.03.006. ISSN 0306-4522.
- ↑ Wells, Samuel A.; Pacini, Furio; Robinson, Bruce G.; Santoro, Massimo (2013). “Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma: An Update”. The Journal of Clinical Endocrinology & Metabolism. 98 (8): 3149–3164. doi:10.1210/jc.2013-1204. ISSN 0021-972X.
- ↑ 16.0 16.1 Image courtesy of Dr Frank Gaillard. Radiopaedia (original file[1]).Creative Commons BY-SA-NC
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Multiple endocrine neoplasia type 2 is caused by a mutation in the RET gene.
Causes
- Mutations in the RET proto-oncogene cause multiple endocrine neoplasia type 2.[1]
- The protein produced by the RET proto-oncogene gene normally plays an important role in signaling cells to respond to their environment, for example by dividing or maturing. Mutations in this gene cause an overactivation of the protein‘s signaling function, which can lead to an overgrowth of cells and the formation of tumors characteristic of multiple endocrine neoplasia type 2.
- MEN2A patients with cutaneous lichen amyloidosis have mutation in codon 634.[2]
- Patients with MEN2A and Hirschsprung disease observed mutations involving RET exon 10.[3]
References
- ↑ Marquard, Jessica; Eng, Charis (September 27, 1999). “Multiple Endocrine Neoplasia Type 2”. GeneReviews® [Internet].
- ↑ Qi, Xiao-Ping; Peng, Jian-Zhong; Yang, Xiao-Wei; Cao, Zhi-Lie; Yu, Xiu-Hua; Fang, Xu-Dong; Zhang, Da-Hong; Zhao, Jian-Qiang (2018). “The RET C611Y mutation causes MEN 2A and associated cutaneous lichen amyloidosis”. Endocrine Connections. 7 (9): 998–1005. doi:10.1530/EC-18-0220. ISSN 2049-3614.
- ↑ Moore, SW; Zaahl, M (2012). “The Hirschsprung’s–multiple endocrine neoplasia connection”. Clinics. 67 (S1): 63–67. doi:10.6061/clinics/2012(Sup01)12. ISSN 1807-5932.
Differentiating Multiple endocrine neoplasia type 2 from other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Multiple endocrine neoplasia type 2 must be differentiated from other hereditary tumors such as medullary thyroid carcinoma, C-cell hyperplasia, pheochromocytoma, von Hippel Lindau syndrome, hereditary paraganglioma–pheochromocytoma, polycythemia and paraganglioma/pheochromocytoma syndrome, neurofibromatosis type 1, and multiple endocrine neoplasia type 1 (MEN 1).
Differential Diagnosis
Multiple endocrine neoplasia type 2 must be differentiated from the following diseases.[1]
| Disease | Gene | Chromosome | Differentiating Features | Components of MEN | Diagnosis | ||
|---|---|---|---|---|---|---|---|
| Parathyroid | Pitutary | Pancreas | |||||
| von Hippel-Lindau syndrome | 3p25.3 |
|
– | – | + |
| |
| Carney complex | 17q23-q24 |
|
– | – | – |
| |
| Neurofibromatosis type 1 | 17 | – | – | – | Prenatal
Postnatal Cardinal Clinical Features” are required for positive diagnosis.
| ||
| Li-Fraumeni syndrome | 17 | Early onset of diverse amount of cancers such as | – | – | – |
Criteria | |
| Gardner’s syndrome | 5q21 |
|
– | – | – |
| |
| Multiple endocrine neoplasia type 2 | – | + | – | – |
Criteria Two or more specific endocrine tumors
| ||
| Cowden syndrome | – | – | – | – |
| ||
| Acromegaly/gigantism | – | – |
|
– | + | – |
|
| Pituitary adenoma | – | – | – | + | – |
| |
| Hyperparathyroidism | – | – | + | – | – |
| |
| Pheochromocytoma/paraganglioma | – | Characterized by | – | – | – |
| |
| Adrenocortical carcinoma |
|
17p, 13q |
|
– | – | – |
|
| Adapted from Toledo SP, Lourenço DM, Toledo RA. A differential diagnosis of inherited endocrine tumors and their tumor counterparts, journal=Clinics (Sao Paulo), volume= 68, issue= 7, 07/24/2013[2] | |||||||
References
- ↑ Toledo, SP; Lourenco Jr, DM; Toledo, RA (2013). “A differential diagnosis of inherited endocrine tumors and their tumor counterparts”. Clinics. 68 (7): 1039–1056. doi:10.6061/clinics/2013(07)24. ISSN 1807-5932.
- ↑ Toledo SP, Lourenço DM, Toledo RA (2013). “A differential diagnosis of inherited endocrine tumors and their tumor counterparts”. Clinics (Sao Paulo). 68 (7): 1039–56. doi:10.6061/clinics/2013(07)24. PMC 3715026. PMID 23917672.
Epidemiology & Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
The prevalence of multiple endocrine neoplasia type 2 is approximately 2.5 per 100,000 individuals worldwide. Multiple endocrine neoplasia type 2B manifests in early infancy while multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma manifest in adulthood.
Epidemiology and Demographics
Prevalence
- Worldwide, the prevalence of multiple endocrine neoplasia type 2 is 2.5 per 100,000 individuals in 2010.[1]
- Multiple endocrine neoplasia type 2 has been reported in approximately 500 to 1000 families worldwide.[2]
- 70-80% of all multiple endocrine neoplasia type 2 cases are multiple endocrine neoplasia type 2A.
Incidence
- In the United States, an estimated 468 cases of multiple endocrine neoplasia type 2-related medullary thyroid cancer are diagnosed per year.[3]
Age
- Multiple endocrine neoplasia type 2 can be found in any age group.
- Multiple endocrine neoplasia type 2B manifests in early infancy while multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma manifest in adulthood.[4]
Gender
- The prevalence and incidence of multiple endocrine neoplasia type 2 do not vary by gender.[5]
Race
- The prevalence of multiple endocrine neoplasia type 2 does not vary by race.[6]
Developed Countries
- In United States the prevalence of multiple endocrine neoplasia type 2 is 2 – 4 per 100,000 individuals.[7]
References
- ↑ Wells, Samuel A.; Pacini, Furio; Robinson, Bruce G.; Santoro, Massimo (2013). “Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma: An Update”. The Journal of Clinical Endocrinology & Metabolism. 98 (8): 3149–3164. doi:10.1210/jc.2013-1204. ISSN 0021-972X.
- ↑ Multiple endocrine neoplasia type2. Orphanet (30.10.2015). http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=653 Accessed on October, 30, 2015
- ↑ Medullary thyroid genetics. National cancer institute (30.10.2015). http://www.cancer.gov/types/thyroid/hp/medullary-thyroid-genetics-pdq#cit/section_1.7 Accessed on October, 30, 2015
- ↑ Wells, Samuel A.; Pacini, Furio; Robinson, Bruce G.; Santoro, Massimo (2013). “Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma: An Update”. The Journal of Clinical Endocrinology & Metabolism. 98 (8): 3149–3164. doi:10.1210/jc.2013-1204. ISSN 0021-972X.
- ↑ Romei, C.; Pardi, E.; Cetani, F.; Elisei, R. (2012). “Genetic and Clinical Features of Multiple Endocrine Neoplasia Types 1 and 2”. Journal of Oncology. 2012: 1–15. doi:10.1155/2012/705036. ISSN 1687-8450.
- ↑ Romei, C.; Pardi, E.; Cetani, F.; Elisei, R. (2012). “Genetic and Clinical Features of Multiple Endocrine Neoplasia Types 1 and 2”. Journal of Oncology. 2012: 1–15. doi:10.1155/2012/705036. ISSN 1687-8450.
- ↑ Norton, Jeffrey A.; Krampitz, Geoffrey; Jensen, Robert T. (2015). “Multiple Endocrine Neoplasia”. Surgical Oncology Clinics of North America. 24 (4): 795–832. doi:10.1016/j.soc.2015.06.008. ISSN 1055-3207.
]]
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Family history is the most common risk factor in the development of multiple endocrine neoplasia type 2.
Risk Factors
- Family history is the most common risk factor in the development of multiple endocrine neoplasia type 2.[1][2][3][4]
- In 1999, during the Seventh International Multiple Endocrine Neoplasia Meeting in Gubbio , the risk of medullary thyroid cancer has been stratified in three categories according to the mutations of c-RET as following.[5][3]
| Gene | Risk |
|---|---|
| Children with multiple endocrine neoplasia type 2B and/or c-RET codon 883, 918,
922 |
Highest risk of aggressive medullary thyroid cancer |
| Children with any c-RET codon 611, 618, 620 or 634
mutations |
High risk of medullary thyroid cancer |
| Children with c-RET codon 609, 768, 790, 791, 804
and 891 mutations |
Less aggressive and slowly growing medullary thyroid cancer |
| Adapted from Marquard J, Eng C. Multiple Endocrine Neoplasia Type 2. 1999 Sep 27 [Updated 2015 Jun 25]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1257/[6] | |
References
- ↑ Yip, Linwah (2003). “Multiple Endocrine Neoplasia Type 2”. Archives of Surgery. 138 (4): 409. doi:10.1001/archsurg.138.4.409. ISSN 0004-0010.
- ↑ Multiple endocrine neoplasia type2. Orphanet (30.10.2015). http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=653 Accessed on October, 30, 2015
- ↑ 3.0 3.1 Marini, Francesca; Falchetti, Alberto; Del Monte, Francesca; Carbonell Sala, Silvia; Tognarini, Isabella; Luzi, Ettore; Brandi, Maria (2006). Orphanet Journal of Rare Diseases. 1 (1): 45. doi:10.1186/1750-1172-1-45. ISSN 1750-1172. Missing or empty
|title=(help) - ↑ Friedhelm Raue & Karin Frank-Raue (2012). “Genotype-phenotype correlation in multiple endocrine neoplasia type 2”. Clinics (Sao Paulo, Brazil). 67 Suppl 1: 69–75. doi:10.6061/clinics/2012(sup01)13. PMID 22584709.
- ↑ Liu, Qiuli; Tong, Dali; Yuan, Wenqiang; Liu, Gaolei; Yuan, Gang; Lan, Weihua; Zhang, Dianzheng; Zhang, Jun; Huang, Zaoming; Zhang, Yao; Jiang, Jun (2017). “Different RET gene mutation-induced multiple endocrine neoplasia type 2A in 3 Chinese families”. Medicine. 96 (3): e5967. doi:10.1097/MD.0000000000005967. ISSN 0025-7974.
- ↑ Jessica Marquard & Charis Eng (1993). “Multiple Endocrine Neoplasia Type 2”. PMID 020301434.
Screening
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
According to the American Society of Clinical Oncology, screening for multiple endocrine neoplasia type 2 by RET gene testing is recommended for children with increased risk of multiple endocrine neoplasia type 2.
Screening
- The DNA-based testing of the c-RET gene is recommended for children with increased risk of multiple endocrine neoplasia type 2.[1][2][3][4]
- The DNA-based testing of the c-RET gene can be easily performed on a blood sample at any age.
- The DNA-based testing of the c-RET gene offers the opportunity for early identification of the c-RET germline mutations, thus contributing to the reduction of morbidity and mortality of multiple endocrine neoplasia type 2 syndrome. In fact, the early recognition of the mutant gene carriers makes possible the prevention and cure of medullary thyroid cancer, by performing a prophylactic thyroidectomy before the clinical expression of the tumor.
- The DNA-based testing of the c-RET gene test is also of importance to detect and thus, to reduce the risk of an unsuspected pheochromocytoma.
- Screening for multiple endocrine neoplasia type 2 include the following tests:
References
|
Natural History, Complications & Prognosis
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Multiple endocrine neoplasia type 2 has a variable natural history. Life threatening complications of multiple endocrine neoplasia type 2 include malignant hypertension, megacolon, and metastasis. Prognosis of multiple endocrine neoplasia type 2 is mainly related to the stage-dependent prognosis of medullary thyroid cancer.
Natural History
Multiple endocrine neoplasia type 2 has a variable natural history. Specific c-RET mutations, are associated to peculiar clinical phenotypes and thus to different courses of the disease. Multiple endocrine neoplasia type 2 can vary from a dormant type after the surgical removal of medullary thyroid carcinoma to a rapidly progressive one with metastatic complications resulting in death.[1]
Complications
- Common complications of multiple endocrine neoplasia type 2 include the following:[2]
Systemic Complications
Endocrine Complications
Genitourinary System Complications
Gastrointestinal System Complications
- Obstruction
- Infection
- Stricture
- Fistula
- Constipation
- Fecal incontinence
- Diverticulum
- Gastrointestinal ganglioneuromatosis
- Megacolon
- Colitis
Prognosis
- Prognosis of multiple endocrine neoplasia type 2 is mainly related to the stage-dependent prognosis of medullary thyroid cancer indicating the necessity of a complete thyroid surgery for index cases with medullary thyroid cancer and the early thyroidectomy for screened at risk subjects.[3]
- The 10 year survival of medullary thyroid cancer of multiple endocrine neoplasia type 2 is 61-76%.[4]
- Multiple endocrine neoplasia type 2A has a better prognosis among the tumors of multiple endocrine neoplasia. Specific c-RET mutations, in fact, are associated to peculiar clinical phenotypes and thus to different course and prognosis of the disease. The following factors influencing survival rate of medullary thyroid carcinoma.
| Stage | Prognosis |
|---|---|
| Stage 1 | Best Prognosis and low chance of recurrence |
| Stage 2 | Good Prognosis and low chance of recurrence |
| Stage 3 | Bad Prognosis and high chance of recurrence |
| Stage 4 | Bad Prognosis and high chance of recurrence |
References
- ↑ Marini, Francesca; Falchetti, Alberto; Del Monte, Francesca; Carbonell Sala, Silvia; Tognarini, Isabella; Luzi, Ettore; Brandi, Maria (2006). Orphanet Journal of Rare Diseases. 1 (1): 45. doi:10.1186/1750-1172-1-45. ISSN 1750-1172. Missing or empty
|title=(help) - ↑ Cohen MS, Phay JE, Albinson C, DeBenedetti MK, Skinner MA, Lairmore TC; et al. (2002). “Gastrointestinal manifestations of multiple endocrine neoplasia type 2”. Ann Surg. 235 (5): 648–54, discussion 654-5. PMC 1422490. PMID 11981210.
- ↑ Raue, Friedhelm; Frank-Raue, Karin (2018). “Update on Multiple Endocrine Neoplasia Type 2: Focus on Medullary Thyroid Carcinoma”. Journal of the Endocrine Society. 2 (8): 933–943. doi:10.1210/js.2018-00178. ISSN 2472-1972.
- ↑ Raue, Friedhelm; Frank-Raue, Karin (2015). “Risk-stratified follow-up of patients with medullary thyroid carcinoma”. International Journal of Endocrine Oncology. 2 (4): 249–252. doi:10.2217/ije.15.23. ISSN 2045-0869.
- ↑ Thyroid cancer: Stage and prognosis. American association of endocrine surgeons (30.10.2015). http://endocrinediseases.org/thyroid/cancer_prognosis.shtml Accessed on October, 30, 2015
- ↑ Roman, Sanziana; Lin, Rong; Sosa, Julie Ann (2006). “Prognosis of medullary thyroid carcinoma”. Cancer. 107 (9): 2134–2142. doi:10.1002/cncr.22244. ISSN 0008-543X.
Diagnosis
Diagnosis
Diagnostic Criteria | History & Symptoms | Physical Examination | Laboratory Findings | Electrocardiogram | X Ray | CT | MRI | Echocardiography or Ultrasound | Other Imaging Findings | Other Diagnostic Studies
Treatment
Treatment
Medical Therapy | Surgery | Primary Prevention | Secondary Prevention | Cost-Effectiveness of Therapy | Future or Investigational Therapies
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH