
Pulmonary hypertension
For patient information click here
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Assistant Editor(s)-in-Chief: José Eduardo Riceto Loyola Junior, M.D.[2], Ralph Matar, Lisa Prior, Ann Slater, R.N., Rim Halaby, Mohamed Moubarak, M.D. [3]
Synonyms and keywords: Hypertensive pulmonary vascular disease; pulmonary arterial hypertension; PAH; hypertensive pulmonary vascular disease; Ayerza syndrome; Ayerza’s syndrome; Ayerza-Arrilaga syndrome
Overview
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1], Richard Channick, M.D.; Assistant Editor(s)-in-Chief: Lisa Prior, Ann Slater, R.N.; José Eduardo Riceto Loyola Junior, M.D.[2]
Overview
Pulmonary hypertension (PH) is a condition of elevated mean blood pressure (mPAP >20 mmHg at rest) within the pulmonary artery of the lungs, leading to a myriad of symptoms including shortness of breath, dizziness, syncope, chest pain, tachycardia and leg swelling. PH has several differing etiologies and is a progressive and fatal disease if left untreated. PH is classified by the European Society of Cardiology and the European Respiratory Society (ESC/ERS 2015) into 5 different groups, which will be discussed later on this page. Treatment is generally targeted toward the underlying etiology of disease. “Pulmonary arterial hypertension” (PAH) refers to group 1 PH in the updated WHO classification and “pulmonary hypertension” (PH) refers to any of group 2 PH through group 5 PH. PH is also used when referring to all five groups collectively.[1]
Historical Perspective
Pulmonary hypertension was first described by Ernst von Romberg, a German physician, in 1891.
Classification
Pulmonary hypertension may be classified according to the mechanism leading to its development into 5 groups: pulmonary arterial hypertension, pulmonary hypertension due to left heart disease, pulmonary hypertension due to chronic lung diseases and/or hypoxia, and pulmonary hypertension due to embolic disease and miscellaneous causes.
| Definition | Characterisitics |
|---|---|
| PH | mPAP >20 mmHg |
| pre-capillary PH | mPAP > 20 mmHg
PCWP ≤ 15 mmHg PVR > 2 WU |
| Isolated post-capillary PH | mPAP > 20 mmHg
PCWP > 15 mmHg PVR ≤ 2 WU |
| Combined post- and pre-capillary PH | mPAP > 20 mmHg
PCWP > 15 mmHg PVR > 2 WU |
| Exercise PH | mPAP/CO slope > 3 mmHg/L/min between rest and exercise |
Pathophysiology
Pulmonary hypertension (PH) is a pathological condition of the pulmonary vasculature present in several disease states that presents with elevated mean pulmonary artery pressure (PAP) as measured by right heart catheterization at rest. The factors that are involved in the pathophysiology of the increase in the mean pulmonary arterial pressure are: increase in pulmonary vascular resistance, increase in the right-side cardiac output, and increase in the mean pulmonary venous pressure. Pulmonary arterial hypertension is characterized by endothelial dysfunction resulting from an imbalance between apoptosis and proliferation of pulmonary artery smooth muscle cells favoring the proliferation. The other types of pulmonary hypertension are caused by hemodynamic changes that result in increased pulmonary blood pressure or architectural changes to the lung or its vasculature that result in the same outcome.
Causes
The World Health Organization (WHO) has classified PH based on etiology into five distinct groups: Group 1 (pulmonary arterial hypertension – which may be related to heritable PAH due to genetic defects, connective tissue diseases such as rheumatoid arthritis, systemic lupus erythematosus, Raynaud Disease, and mixed connective tissue disease, HIV, drugs and toxins, and parasitic infections such as schistosomiasis), Group 2 (PH due to left heart failure), Group 3 (PH due to chronic lung disease and/or hypoxemia), Group 4 (PH due to chronic thromboembolic disease), and Group 5 (PH due to multifactorial mechanisms such as chronic hemolytic anemia, sarcoidosis, chronic kidney disease, and myeloproliferative disorders).
Differentiating Pulmonary hypertension from Other Diseases
One of the most common initial presentations of patients with pulmonary hypertension is dyspnea; therefore, the differential diagnosis is very broad. As the disease progresses with time, more symptoms related to right ventricular hypertrophy and failure occur; which further narrows down the differential diagnosis.
Epidemiology and Demographics
Pulmonary arterial hypertension has been considered as a disease of young women. The mean age of patients in the U.S. registry was 36 years old and the overall female-to-male ratio was 1.7:1. The prevalence of pulmonary hypertension is approximately 1.5-5 per 100,000 individuals.
Risk Factors
Pulmonary hypertension (PH) is a multifactorial disease involving genetic and environmental risk factors. Risk factors for pulmonary arterial hypertension include BMPR2 mutation, connective tissue disease, HIV infection, portal hypertension, fenfluramine use, and congenital heart disease with shunt. Left heart and lung diseases are risk factors for PH. Patients with a hypercoagulable state (such as the presence of lupus anticoagulant, deficiency of protein C, protein S, or antithrombin III, chronic inflammatory disorders, myeloproliferative syndromes, and splenectomy) are at an increased risk for chronic thromboembolic pulmonary hypertension.
Screening
Patients with a known BMPR2 mutation, scleroderma, and portal hypertension undergoing evaluation for liver transplantation should receive periodic screening for pulmonary hypertension (PH) through a thorough assessment of the presence of symptoms, physical examination, a chest X ray, electrocardiography, and an echocardiogram. Additional investigation with right heart catheterization should be performed if screening is suggestive of the presence of PH.
Natural History, Complications and Prognosis
The most common initial symptoms of pulmonary hypertension are dyspnea, fatigue, and syncope. There was an estimated median survival of 2.8 years for symptomatic patients who do not receive any treatment, with the most common cause of death as cor pulmonale, but survival rates have been increasing as new forms of treatment become available. Despite such advances, prognosis is still poor.
Diagnosis
Diagnostic Study of Choice
Pulmonary hypertension is defined by a mean pulmonary arterial pressure higher than 25mmHg. It can be assessed by echocardiography, the diagnostic study of choice due to its low risk and useful information that it can provide, and right heart cardiac catheterization to confirm the diagnosis.
History and Symptoms
The hallmark of pulmonary hypertension is progressive dyspnea. The most common symptoms of pulmonary hypertension include dyspnea, fatigue, chest pain and syncope or presyncope. Ortner syndrome may also be seen (characterized by hoarseness due to compression of the left laryngeal nerve caused by enlargement of the pulmonary artery).
Physical Examination
Pulmonary hypertension (PH) can present with myriad physical findings that may be associated with PH or the underlying cause. Findings associated with pulmonary hypertension are usually associated with right heart failure or right heart overload, while other findings may be associated with underlying cause, such as thoracic deformities which may arise in the setting of COPD, or sclerodactyly that may be seen in patients with scleroderma. Some may develop on a spectrum corresponding to the severity of the disease.
Laboratory Findings
There are no specific diagnostic lab findings associated with pulmonary hypertension. Despite that, several laboratory tests are required in the evaluation of a patient for pulmonary hypertension to assess its severity or possible associated causes. Biochemistry, hematology and thyroid function tests are required in all patients with pulmonary hypertension. They are important for the diagnosis of chronic hemolytic anemia, myeloproliferative disorders, thyroid disorders and chronic renal failure on dialysis.
ECG
Elevated pulmonary pressures in pulmonary hypertension (PH) can lead to right ventricular hypertrophy (RVH) and right atrial enlargement which can sometimes be observed on an electrocardiogram (ECG). The ECG findings of PH include right axis deviation, right ventricular strain pattern, and P pulmonale. The ECG findings of PH are neither specific nor sensitive and their absence does not rule out the presence of PH.
Chest X Ray
A chest X-ray is abnormal in the majority of patients with pulmonary hypertension (PH); however, there is no correlation between the severity of PH and the findings on a chest X-ray. Findings of PH on a chest X-ray include pulmonary artery dilatation and right-sided enlargement of the heart. A chest X-ray allows the exclusion of left heart disease and lung disease that can lead to group 2 and group 3 PH, respectively.
CT
A lung CT scan is helpful in the differential diagnosis of pulmonary hypertension. Different types of CT imaging have been used to rule out certain etiologies of pulmonary hypertension and to evaluate the anatomy of the pulmonary vasculature.
MRI
Thoracic MRI is helpful in the differential diagnosis of pulmonary hypertension as well as in the evaluation of the right ventricle function. It provides important prognostic indicators when assessing the right ventricle in patients with pulmonary hypertension. Findings that predict poor prognosis are: stroke volume ≤25ml/m^2, right ventricular end-diastolic volume ≥84ml/m^2 and left ventricular end-diastolic volume ≤40ml/m^2.
Echocardiography or Ultrasound
Echocardiography may demonstrate right ventricular or atrial enlargement with a thickened interventricular septum in patients with pulmonary hypertension, decreased right ventricular function, or hypertrophy. Pulmonary artery systolic pressure can also be estimated using echocardiography. Right ventricular afterload may be suggested by a leftward septal displacement during systole. Pericardial effusions and diminished left ventricular cavity typically portend a dismal prognosis.
Other diagnostic studies
Pulmonary hypertension diagnosis is made using right heart cardiac catheterization. It is mandatory for diagnosing pulmonary arterial hypertension. It confirms the diagnosis and evaluates for some causes such as valvular heart diseases. Other studies may also be performed. These studies include pulmonary function tests, overnight oximetry, and ventilation-perfusion studies (which is crucial to rule out chronic pulmonary thromboembolism).
Treatment
Medical Therapy
The choice of treatment for pulmonary hypertension (PH) requires the assessment of the clinical severity of the disease and the identification of any underlying cause. Patients who have PH secondary to a medical condition such as left heart failure, lung diseases, or thromboembolic disease (PH group 2, 3, and 4 respectively) should receive treatment for the underlying cause. Patients who have pulmonary arterial hypertension (PAH) must undergo vasoreactivity testing in order to assist in the selection of the optimal therapy which include calcium channel blockers, endothelin receptor antagonist, phosphodiesterase inhibitors, or prostanoids.
Surgery
Patients with severe WHO functional class II or III pulmonary hypertension (PH) refractory to medical therapy are candidates for surgical intervention, such as atrial septostomy or lung transplantation. Pulmonary thromboendarterectomy (PTE) is a surgical procedure that is used for chronic thromboembolic pulmonary hypertension.
Primary Prevention
Genetic and environmental factors are involved in pulmonary hypertension (PH); therefore, not all cases of PH are preventable. PH that is secondary to other diseases such as left heart failure, chronic lung disease, chronic liver disease, and collagen vascular diseases among others can be prevented by the early and optimal treatment of these medical conditions. Patients who are at elevated risk for developing pulmonary arterial hypertension (PAH) must be monitored for the occurrence of symptoms of PAH. Patients at risk for PAH include subjects with systemic sclerosis or with genetic predisposition.
Secondary Prevention
The recommended measures for the secondary prevention are avoiding pregnancy, rigorous follow up in case of pregnancy, avoid unnecessary surgeries, multidisciplinary care in case of necessary surgery, avoid high altitude, supplemental oxygen in order to ensure a target oxygen saturation of 91% in case of exposure to high altitude, and up-to-date immunizations against influenza and pneumococcal pneumonia.
References
Historical Perspective
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]: Associate Editor(s)-in-Chief: José Eduardo Riceto Loyola Junior, M.D.[2]
Overview
Pulmonary hypertension was first described by Ernst von Romberg, a German physician, in 1891.
Historical Perspective
- Pulmonary hypertension was first described by Ernst von Romberg, a German physician, in 1891.
- In 1929, Werner Forssman demonstrated that it was possible to perform right-sided catheterization in humans by performing catheterization on himself.
- In 1951, Dresdale coined the term primary pulmonary hypertension after describing series of cases of this new pathology. He also investigated the effects of tolzoline in a woman with pulmonary arterial hypertension causing a sudden reduction in pulmonary vascular resistance.
- In the late 1960s, there was an epidemic of pulmonary arterial hypertension induced by aminorex, which sparked interest in the disease.
- Pulmonary hypertension was first classified into primary and secondary in 1973 during the World Health Organization (WHO) meeting on PH in Geneva, Switzerland.
- Bosentan was approved to treat pulmonary hypertension in 2001, and sildenafil was approved in 2005.[1]
References
- ↑ Barst RJ (2008). “Pulmonary hypertension: past, present and future”. Ann Thorac Med. 3 (1): 1–4. doi:10.4103/1817-1737.37832. PMC 2700428. PMID 19561874.
Classification
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1], Richard Channick, M.D.; Assistant Editor(s)-in-Chief: Ralph Matar; Lisa Prior, Ann Slater, R.N.; Rim Halaby, M.D. [2]; José Eduardo Riceto Loyola Junior, M.D.[3]
Overview
Pulmonary hypertension may be classified according to the mechanism leading to its development into 5 groups: pulmonary arterial hypertension, pulmonary hypertension due to left heart disease, pulmonary hypertension due to chronic lung diseases and/or hypoxia, and pulmonary hypertension due to embolic disease, and miscellaneous causes.
Classification
- Pulmonary hypertension was first classified into primary and secondary in 1973 during the World Health Organization (WHO) meeting on PH in Geneva, Switzerland.[1]
- Pulmonary hypertension can be classified following different methods such as using the WHO clinical criteria, the hemodynamic findings, and the histopathological findings. The most common method of classification is using the disease mechanism, established by the World Health Organization, which is discussed below in detail.
WHO – Clinical Classification
- Pulmonary hypertension was first classified into primary and secondary in 1973 during the World Health Organization (WHO) meeting on PH in Geneva, Switzerland.[1]
- The classification of the disease has been progressively updated since then and the latest version was defined in 2018, during the 6th World Symposium on Pulmonary Hypertension.
- It is currently used by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the labeling of new drugs approved for the treatment of pulmonary hypertension.
- The latest classification method categorizes pulmonary hypertension into 5 groups:
- Group I – Pulmonary arterial hypertension
- Group II – Pulmonary hypertension due to left heart disease
- Group III – Pulmonary hypertension due to chronic lung diseases and/or hypoxia
- Group IV – Pulmonary hypertension due to embolic disease
- Group V – Miscellaneous causes (e.g., sarcoidosis, lymphatic obstruction)
WHO Classification
Shown below is a table with the detailed classification of pulmonary hypertension.[2]
Abbreviations: BMPR, bone morphogenic protein receptor type II; CAV1, caveolin-1; ENG, endoglin; HIV, human immunodeficiency virus.
| Group 1. Pulmonary arterial hypertension (PAH) |
| 1.1. Idiopathic PAH |
| 1.2. Heritable PAH 1.2.1 BMPR2 |
| 1.3 Drug and toxin-induced Definite (an epidemic or large multicenter epidemiological studies demonstrating an association between a drug and PAH) Likely (a single case-control study demonstrating an association or a multiple-case series) Possible (drugs with similar mechanisms of action as those in the definite or likely category but which have not yet been studied)
Unlikely (one in which a drug has been studied in epidemiological studies and an association with PAH has not been demonstrated) |
| 1.4 Associated with: 1.4.1 Connective tissue disease |
| 1’ Pulmonary veno-occlusive disease (PVOD) and/or pulmonary capillary hemangiomatosis (PCH) |
| 1’’ Persistent pulmonary hypertension of the newborn (PPHN) |
| Group 2. Pulmonary hypertension due to left heart disease |
| 2.1 Left ventricular systolic dysfunction |
| 2.2 Left ventricular diastolic dysfunction |
| 2.3 Valvular disease |
| 2.4 Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies |
| Group 3. Pulmonary hypertension due to lung diseases and/or hypoxia |
| 3.1 Chronic obstructive pulmonary disease |
| 3.2 Interstitial lung disease |
| 3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern |
| 3.4 Sleep-disordered breathing |
| 3.5 Alveolar hypoventilation disorders |
| 3.6 Chronic exposure to high altitude |
| 3.7 Developmental lung diseases |
| Group 4. Chronic thromboembolic pulmonary hypertension (CTEPH) |
| Group 5. Pulmonary hypertension with unclear multifactorial mechanisms |
| 5.1 Hematologic disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy |
| 5.2 Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis |
| 5.3 Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders |
| 5.4 Others: tumor obstruction, fibrosing mediastinitis, chronic renal failure, segmental PH |
Classification Based on Hemodynamical Findings
Abbreviations: PAP: Pulmonary artery pressure; PWP: pulmonary wedge pressure
| Type of pulmonary hypertension | Possible clinical class | Mean PAP | PWP |
| Pre-capillary | Class I Class III Class IV Class V |
≥ 25 mmHg | ≤ 15 mmHg |
| Post-capillary | Class II | ≥ 25 mmHg | > 15 mmHg |
Classification Based on Histopathological Findings
PH is a pathological condition present in different disease states that share similar clinical manifestations and some common histopathological features. Shown below is a table that summarizes the classification of PH based on histopathology findings.[3]
| Class | Histopathological findings[3] |
| Pulmonary arteriopathy | Constrictive lesions in pulmonary arteries:
Complex lesions in pulmonary arteries:
|
| Pulmonary arteriopathy with venous-venular changes | Changes similar to pulmonary arteriopathy PLUS Changes in venules and veins |
| Pulmonary occlusive venopathy (with or without arteriopathy) |
Changes in venules and veins:
Changes in the capillaries:
Changes in the interstitium |
| Pulmonary microvasculopathy (with or without arteriopathy and/on venopathy) |
Changes in the capillaries:
Changes in the interstitium |
| Unclassified | Non specific changes |
References
- ↑ 1.0 1.1 Hatano S, Strasser T. Primary Pulmonary Hypertension. Report on a WHO Meeting. October 15–17, 1973, Geneva: World Health Organization, 1975.
- ↑ Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A; et al. (2013). “Updated clinical classification of pulmonary hypertension”. J Am Coll Cardiol. 62 (25 Suppl): D34–41. doi:10.1016/j.jacc.2013.10.029. PMID 24355639.
- ↑ 3.0 3.1 Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM; et al. (2004). “Pathologic assessment of vasculopathies in pulmonary hypertension”. J Am Coll Cardiol. 43 (12 Suppl S): 25S–32S. doi:10.1016/j.jacc.2004.02.033. PMID 15194175.
Pathophysiology
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Assistant Editor(s)-in-Chief: Ralph Matar; Rim Halaby José Eduardo Riceto Loyola Junior, M.D.[2]
Overview
Pulmonary hypertension (PH) is a pathological condition of the pulmonary vasculature present in several disease states that presents with elevated mean pulmonary artery pressure (PAP) as measured by right heart catheterization at rest. The factors that are in involved in the pathophysiology of the increase in the mean pulmonary arterial pressure are: increase in pulmonary vascular resistance, increase in the right-side cardiac output and increase in the mean pulmonary venous pressure. Pulmonary arterial hypertension is characterized by endothelial dysfunction resulting from an imbalance between apoptosis and proliferation of pulmonary artery smooth muscle cells favoring the proliferation. The other types of pulmonary hypertension are caused by hemodynamic changes that result in increased pulmonary blood pressure or architectural changes to the lung or its vasculature that result in the same outcome.
Pathophysiology
The pathophysiology of pulmonary hypertension[1][2][3][4][5][6][7][8][9][10]
PH is defined as an elevated mean pulmonary artery pressure (PAP) ≥ 25 mmHg as measured by right heart catheterization at rest. The elevation in PAP results from an elevation in the pulmonary vascular resistance caused by a multifactorial pathogenesis involving genetic and environmental factors.
Pulmonary hypertension has several pathophysiologic mechanisms depending on the underlying etiology. Nevertheless, the following sequence of events is almost always present:
- Vasoconstriction (ocurring early in the disease);
- Thrombosis (during the evolution of the disease);
- Remodeling (then takes place and becomes the most important factor).[3]
- An initiating factor leads to increased resistance in the pulmonary vasculature causing narrowing of the vessels and impaired blood flow.
- As a consequence, the right ventricle adapts by increasing right ventricular systolic pressures to preserve the cardiac output from the right heart.
- Over time, increasing right ventricular systolic pressures will subsequently result in chronic changes in the pulmonary circulation and the affected blood vessels progressively become stiffer and thicker, further increasing the blood pressure within the lungs and impairing blood flow.
- In addition, the increased workload of the heart causes thickening and enlargement of the right ventricle, making the heart less able to pump blood through the lungs, causing right heart failure. Factors that might affect the ability of the right ventricle to adapt to an increased pulmonary vascular resistance are:
- Age of the patient at onset
- Rapidity of onset of pulmonary hypertension
- Coexisting hypoxemia
Pulmonary Arterial Hypertension
Molecular and Cellular Changes
- Pulmonary arterial hypertension is characterized by endothelial dysfunction resulting from an imbalance between apoptosis and proliferation of pulmonary artery smooth muscle cells favoring the proliferation.
- There is also thickened and disordered adventitia due to excessive amounts of adventitial metalloproteinases.
- Several pathways are implicated in development of vascular remodeling, which is the hallmark of pathologic changes in pulmonary hypertension:
1. Vasoconstriction-mediated remodeling
- Imbalance of vasoactive signals is suggested in the pathogenesis of remodeling.
- Reduced levels of vasodilatory mediators, in particular prostaglandin I2, nitric oxide, and cyclic guanine monophosphate, and elevated levels of vasocontricting molecules such as endothelin 1, thromboxane, and 5-hydroxytryptamine contribute to alterations in the vascular tone.
- Down regulation of the voltage-gated potassium channels has also been linked with altered pulmonary vascular tone, dysregulation of cellular homeostasis, and induction of proliferative sequelae in vascular smooth muscle cells.
- Patients with PAH seem to have higher concentrations of serum endothelin-1 (a potent vasoconstrictor) than controls.[3]
- May occur in response to hypoxia;
- May be related to calcium channel dysfunction – in fact, there are mutations in the ion channels (KCNK3) which predispose to hereditary PAH;
- May be reversible – patients treated early in the disease with calcium channel blockers seem to have a much better prognosis.[3]
2. Thrombosis-mediated remodeling
- Many coagulopathies were described in patients with PAH, such as increased von Willebrand factor activity, protein C and protein S deficiency.[11][12]
- Thrombin plays a key role in mediating vascular remodeling by activating platelet, upregulating angiogenesis-related genes (including tissue factor [TF], basic fibroblast growth factor [bFGF], and matrix metalloproteinase-2), and transactivating vascular endothelial growth factor (VEGF) by inducing the production of reactive oxygen species (ROS) and the expression of the hypoxia inducible factor-1 alpha.
- Upregulation of TF in the vasculature leads to initiation of coagulation cascade and migration and proliferation of smooth muscle cells upon vascular injury.
- In addition, platelet activation by thrombin results in the release of granules containing substances that promote mitogenesis and vasoconstriction including VEGF, bFGF, platelet-derived growth factor, and serotonin, which also contribute to increased endothelial cell migration and proliferation.
3. Proliferation-mediated remodeling
- The pathogenic pathway of lung vasculature remodeling in PH is not fully understood;
- It is regulated by a variety of growth factors, cytokines, mytogens, ion channels, neurotransmitters viruses, transcription factors and receptors, the most studied being the bone morphogenetic protein receptor type II (BMPR2).
- BMPR2 loss-of-function mutations were found in 70% of patients with hereditary PAH and in 20% of patients with idiopathic PAH;[13][14]
- Anecdotal reports have stated that imatinib has been able to cause significant improvement of PAH patients;[15]
- Serotonin exerts both vasoconstrictive and mitogenic effects on smooth muscle cells (SMC).
- Serotonin m bind to 5-hydroxytryptamine 1A and 2B receptors or may enter SMC via serotonin transporter which induces generation of ROS, rho kinase, and mitogen-activated protein kinases.
- This in turn leads to the expression of growth factors and proliferation. Bone morphogenetic protein (BMP) is also involved in the SMC proliferation.
- Upon binding of BMP to its heterodimerized receptors, a group of cytoplasmic proteins known as receptor-mediated Smads are phosphorylated and translocated to the nucleus where they upregulate genes related to anti-proliferation.
- Serotonin is shown to antagonize the BMP/Smad pathway thus facilitating the proliferation of smooth muscle cells.
4. Inflammation-mediated remodeling
- Influx of inflammatory effector cells is stimulated by the release of chemokines such as CCL5 and CX3CL1.
- On the other hand, endothelial cell and smooth muscle cell dysfunction contributes to the release of vasomotor and growth factors, activation of transcription factors, influx of calcium, and dysfunction of mitochondria.
- The net effect is a shift of balance in favor of proliferation and suppressed apoptosis, leading to remodeling of the pulmonary vasculature.
Shown below is an image depicting remodeling in pulmonary arterial hypertension.

Genetic Mutations
TGF-β Receptor Pathway
- The bone morphogenetic protein receptor II (BMPR2) gene encodes a serine/threonine kinase that functions as a receptor for bone morphogenetic proteins which forms a heterodimer cytoplasmic complex of two type II and two type I receptors. The downstream signaling involves activation of SMAD transcription factor which mediates anti-proliferation in smooth muscle cells.
- Activin-like kinase 1 gene mutations are detected in patients with hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension. Mutation would cause growth-promoting alterations.
Serotoninergic Pathway
- 5- Hydroxytryptamine (serotonin) transporter mutations have also been associated with the proliferation of smooth muscle cells of pulmonary arteries.[16]
Genetics
Familial PAH often results from a mutation in the bone morphogenic protein receptor-2 (BMPR2) and is inherited as an autosomal dominant disease with incomplete penetrance and anticipation.[17]
Associated conditions
- PAH is also associated with:[18]
- Congenital heart disease (30% of untreated cases)
- Connective tissue diseases (12% of patients with scleroderma and up to 21% of patients with rheumatoid arthritis)
- HIV (0.5%)
- Portal hypertension (2-6%)
- Sickle cell disease (20 to 40%)
- Systemic lupus erythematosus (4 to 14%)
- Hemoglobinopathies
- Myeloproliferative disorders
- Drugs and toxins
Histopathology
PH is a pathological condition present in different disease states that share similar clinical manifestation and some common histopathological features. Shown below is a table that summarizes the classification of PH based on histopathology findings.[19]
| Class | Histopathological findings[19] | Images |
|---|---|---|
| Pulmonary arteriopathy | Constrictive lesions in pulmonary arteries
Complex lesions in pulmonary arteries
|
   |
| Pulmonary arteriopathy with venous-venular changes | Changes similar to pulmonary arteriopathy PLUS Changes in venules and veins |
  |
| Pulmonary occlusive venopathy (with or without arteriopathy) |
Changes in venules and veins
Changes in the capillaries
Changes in the interstitium |
|
| Pulmonary microvasculopathy (with or without arteriopathy and/on venopathy) |
Changes in the capillaries
Changes in the interstitium |
  |
| Unclassified | Non specific changes |
Gallery
The images below are courtesy of Dr. Yale Rosen (Source:Wikimedia Commons).
-
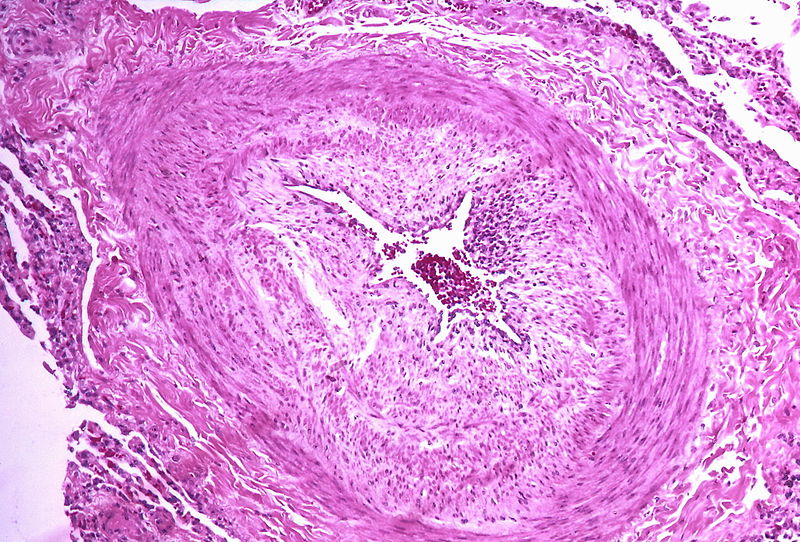 Marked intimal and medial thickening
Marked intimal and medial thickening -
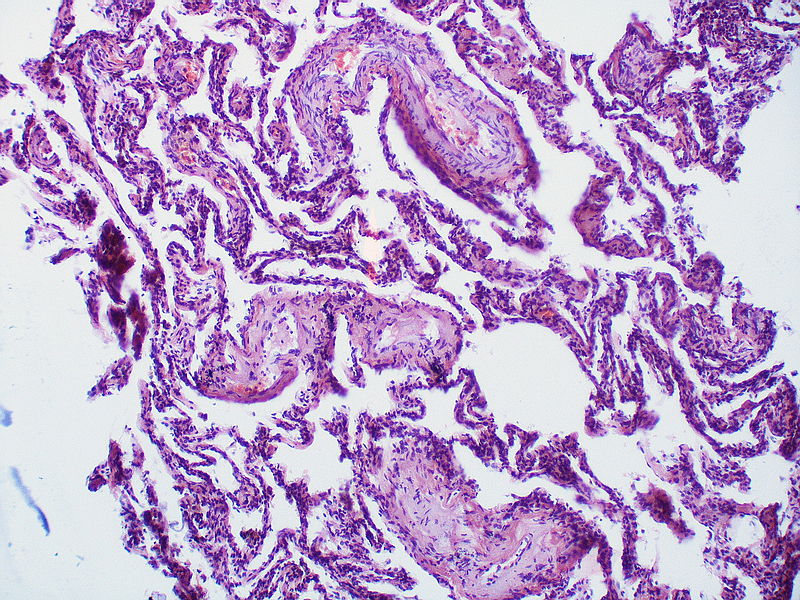 Medial muscle layer thickening involving multiple pulmonary arteries.
Medial muscle layer thickening involving multiple pulmonary arteries. -
This arteriole exhibits fibrinoid necrosis.
-
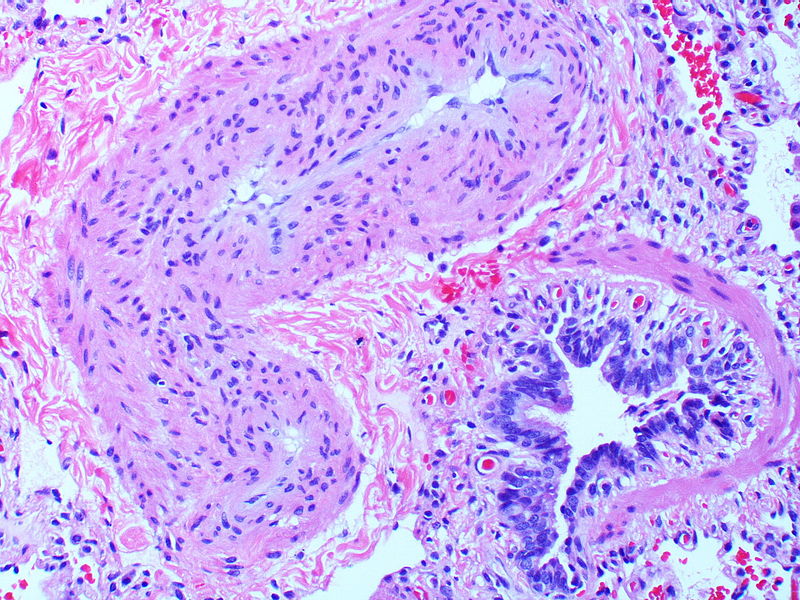 Pronounced thickening of the medial muscle layer.
Pronounced thickening of the medial muscle layer. -
Marked intimal and medial thickening.
-
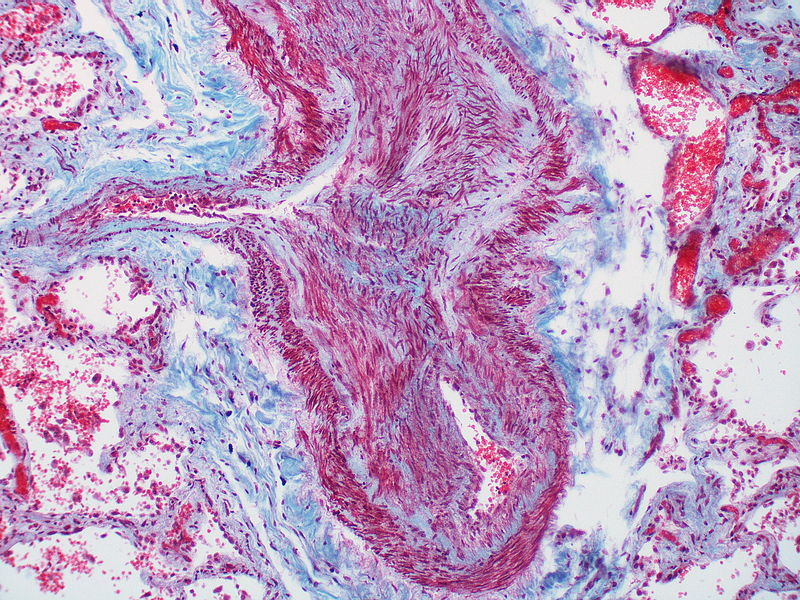 This artery exhibits marked intimal thickening The red-staining cells in the intima are probably myofibroblasts. Masson trichrome stain.
This artery exhibits marked intimal thickening The red-staining cells in the intima are probably myofibroblasts. Masson trichrome stain. -
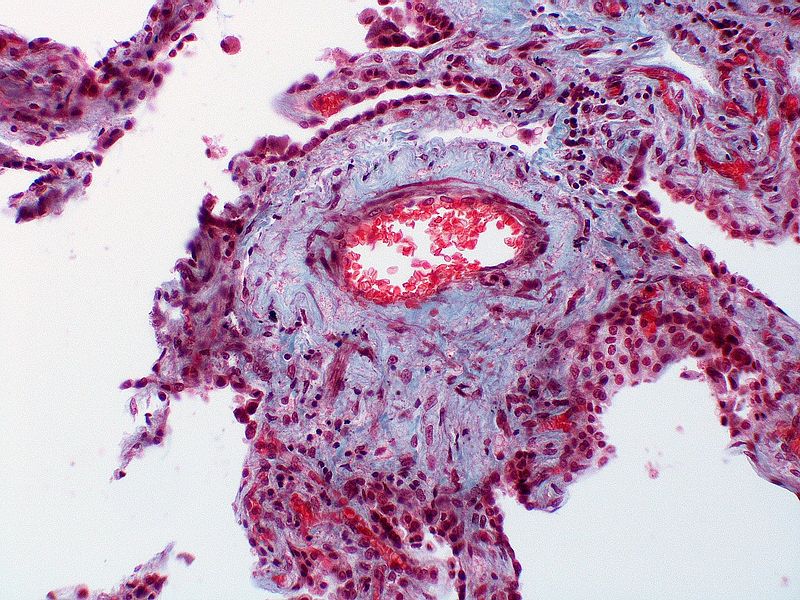 Marked adventitial thickening. Masson trichrome stain.
Marked adventitial thickening. Masson trichrome stain. -
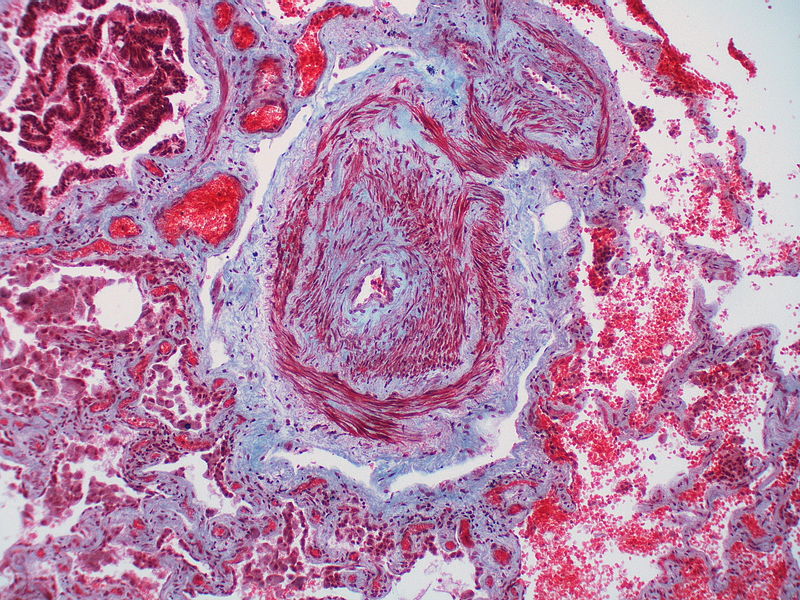 This artery exhibits marked intimal thickening as well as adventitial thickening. The red-staining cells in the intima are probably myofibroblasts. Masson trichrome stain.
This artery exhibits marked intimal thickening as well as adventitial thickening. The red-staining cells in the intima are probably myofibroblasts. Masson trichrome stain. -
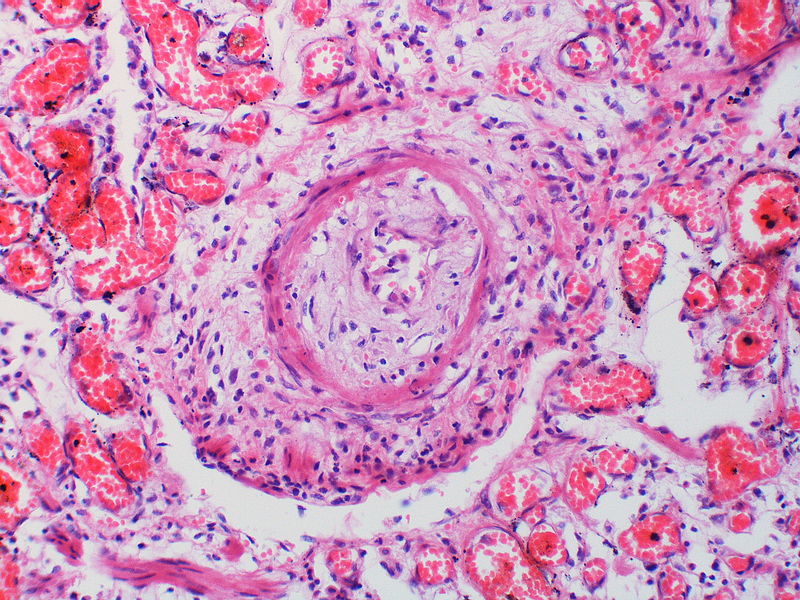 Pulmonary hypertension associated with pulmonary hemangiomatosis. A muscular pulmonary artery surrounded by angiomatous vessels exhibits marked intimal and adventitial thickening.
Pulmonary hypertension associated with pulmonary hemangiomatosis. A muscular pulmonary artery surrounded by angiomatous vessels exhibits marked intimal and adventitial thickening. -
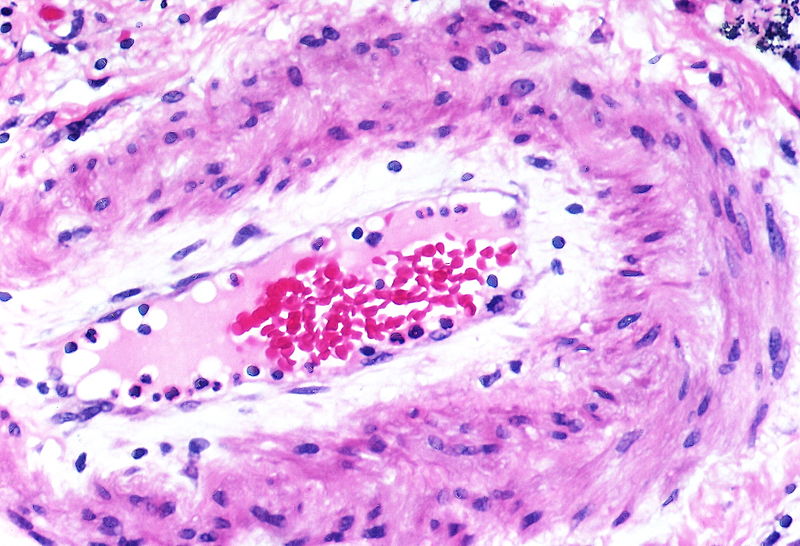 There is thickening of the medial muscle layer as well as mild intimal thickening.
There is thickening of the medial muscle layer as well as mild intimal thickening. -
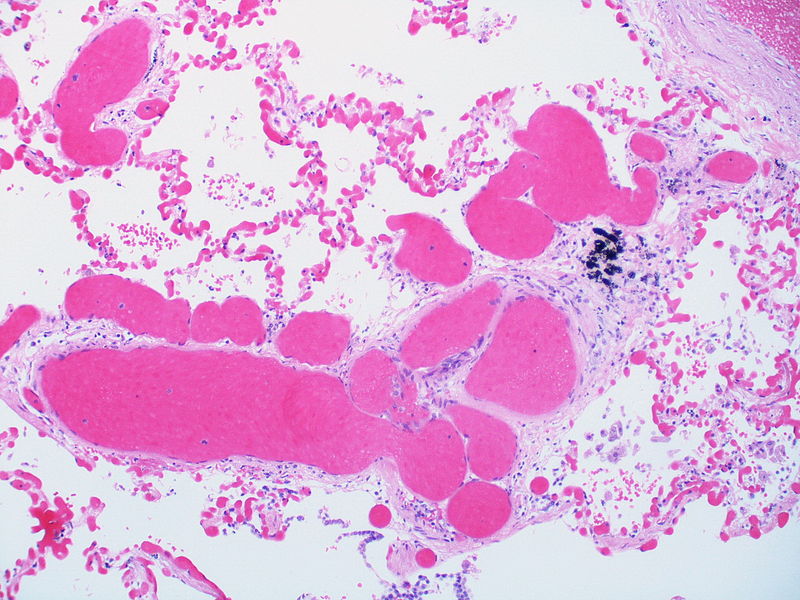 Alveolar capillary proliferation as well as the proliferation of larger blood vessels, probably venules.
Alveolar capillary proliferation as well as the proliferation of larger blood vessels, probably venules. -
This artery exhibits vasculitis characterized by focal necrosis of the wall accompanied by inflammation.
-
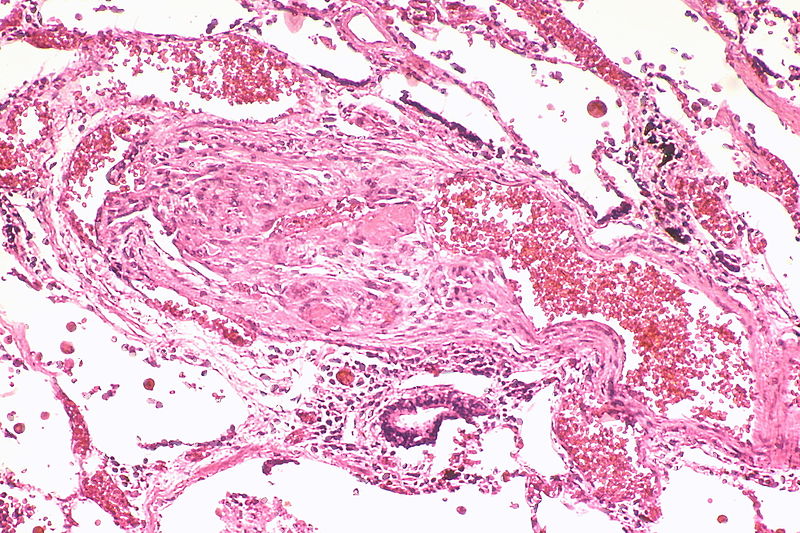 A large dilatation lesion is present on the right side of this image adjacent to an angiomatoid lesion.
A large dilatation lesion is present on the right side of this image adjacent to an angiomatoid lesion. -
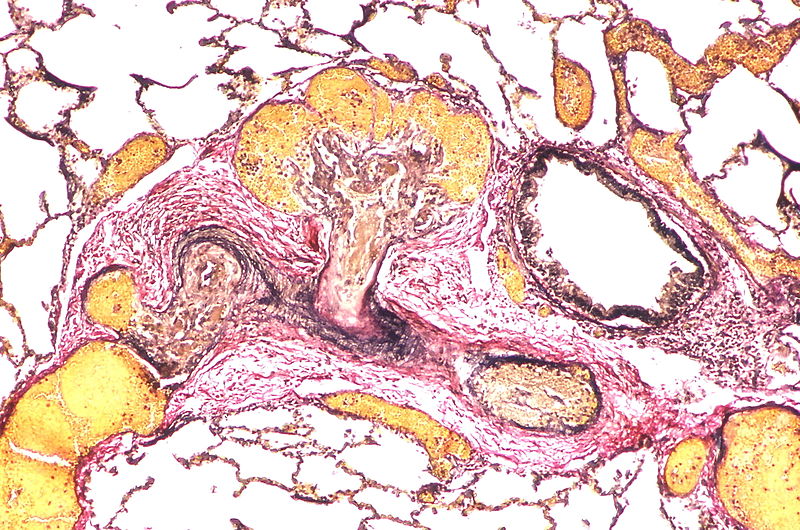 Two angiomatoid lesions are present. A large dilatation lesion is a present adjacent to the angiomatoid lesion that is located just above center.
Two angiomatoid lesions are present. A large dilatation lesion is a present adjacent to the angiomatoid lesion that is located just above center. -
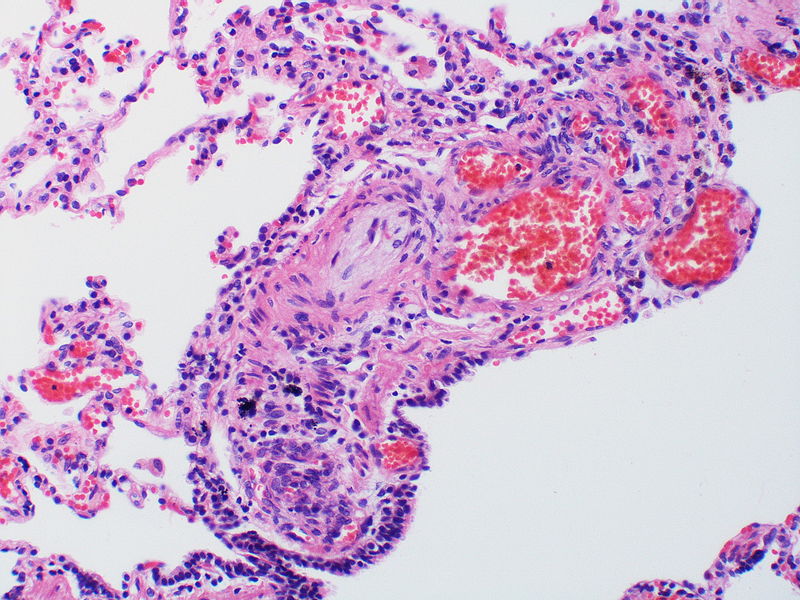 There is angiomatoid (plexiform) lesion.
There is angiomatoid (plexiform) lesion. -
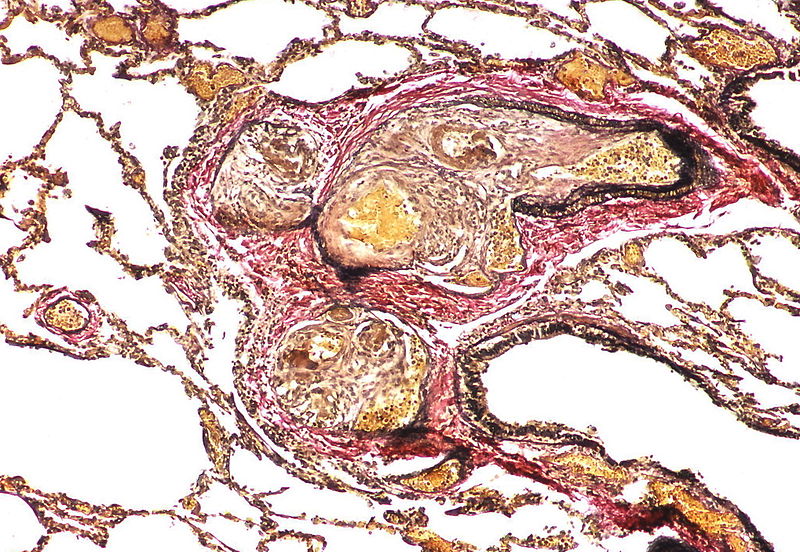 The appearance suggests multiple lesions but a single lesion cannot be excluded. Note the destruction of the arterial wall at the site of the angiomatoid lesion. Destructive arterial changes are frequently associated with angiomatoid lesions. There is an elastic stain.
The appearance suggests multiple lesions but a single lesion cannot be excluded. Note the destruction of the arterial wall at the site of the angiomatoid lesion. Destructive arterial changes are frequently associated with angiomatoid lesions. There is an elastic stain. -
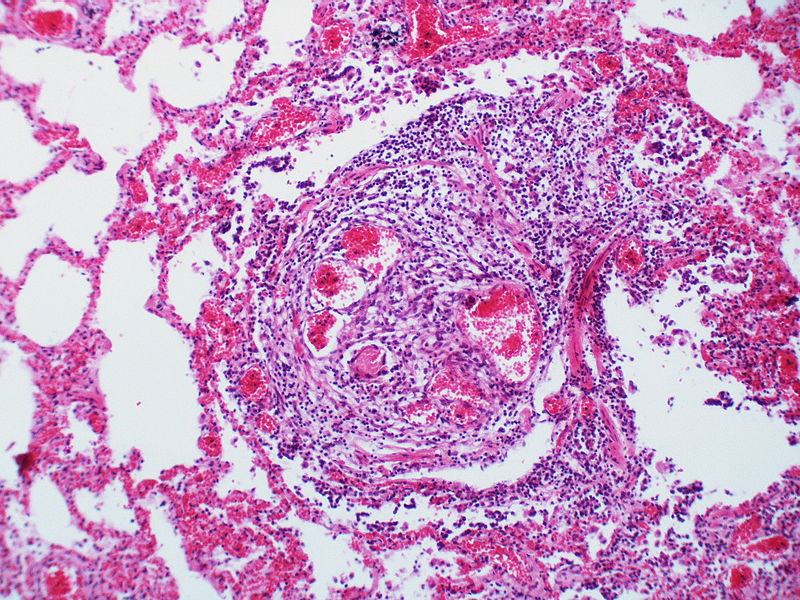 This angiomatoid lesion is accompanied by inflammation which is frequently associated with these lesions.
This angiomatoid lesion is accompanied by inflammation which is frequently associated with these lesions. -
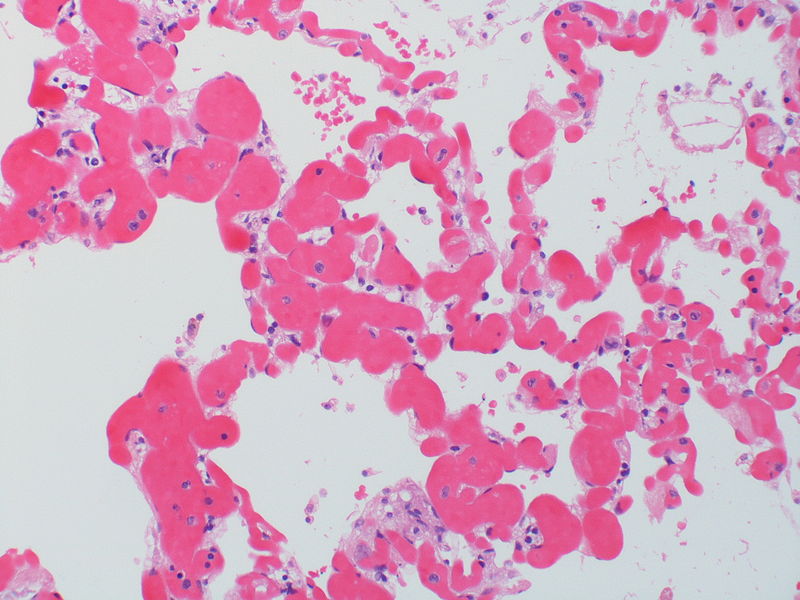 There is a proliferation of alveolar wall capillaries which are markedly dilated.
There is a proliferation of alveolar wall capillaries which are markedly dilated. -
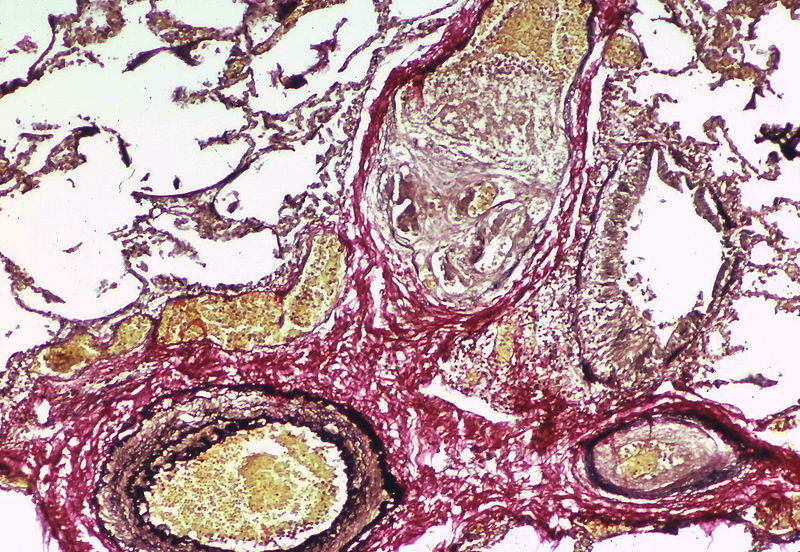 A dilatation lesion is seen in the left lower quadrant adjacent to the angiomatoid lesion. There is an elastic stain.
A dilatation lesion is seen in the left lower quadrant adjacent to the angiomatoid lesion. There is an elastic stain. -
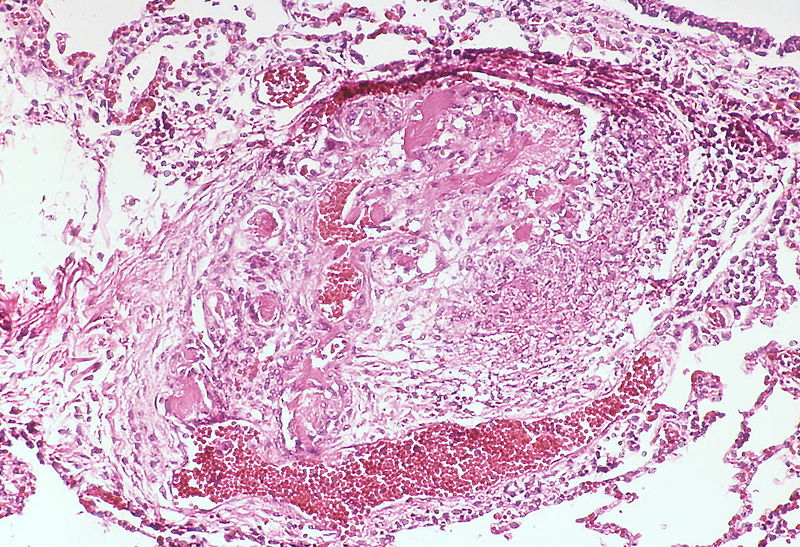 This angiomatoid lesion contains multiple thrombi within the vascular spaces. A large dilatation lesion is a present adjacent to the bottom of the angiomatoid lesion.
This angiomatoid lesion contains multiple thrombi within the vascular spaces. A large dilatation lesion is a present adjacent to the bottom of the angiomatoid lesion. -
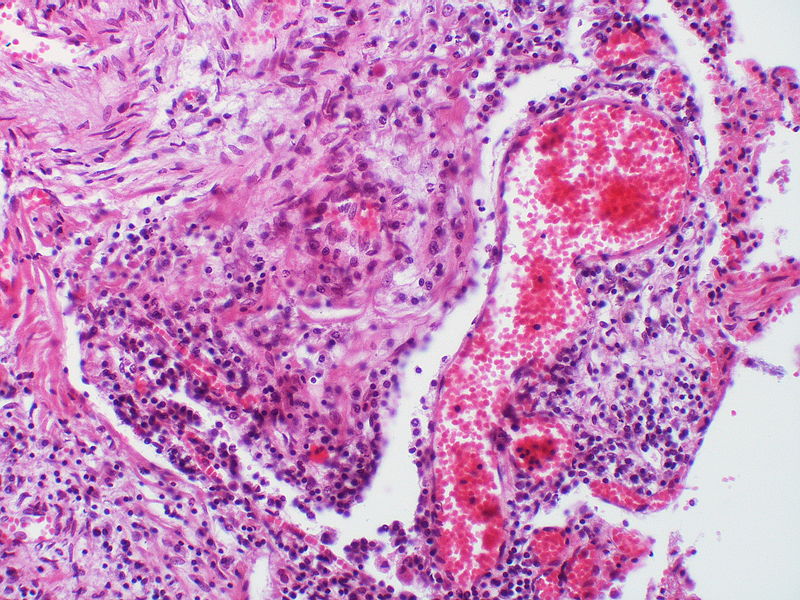 A dilatation lesion is present adjacent to the right side of the angiomatoid lesion.
A dilatation lesion is present adjacent to the right side of the angiomatoid lesion. -
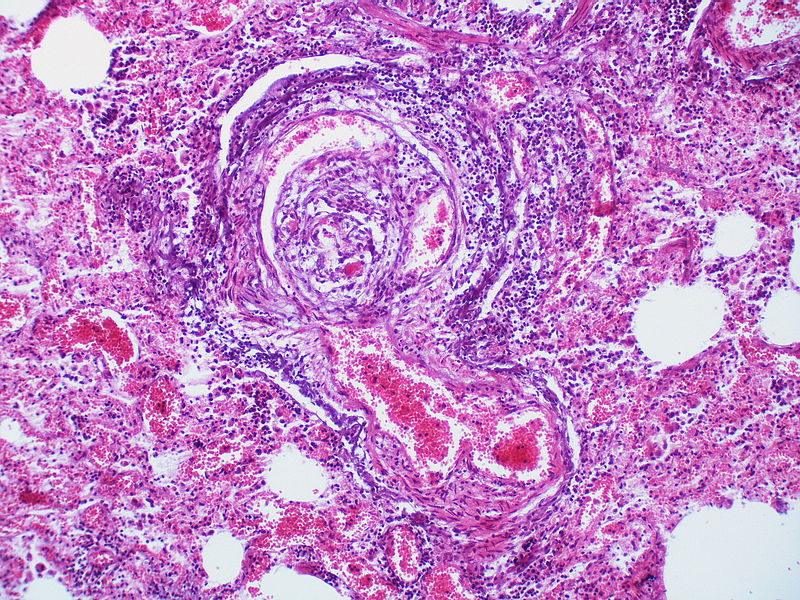 There is a disappearance of the arterial wall at the site of the angiomatoid lesions. Arterial wall destruction is a frequent finding in association with angiomatoid lesions. A dilatation lesion encircles the angiomatoid lesion
There is a disappearance of the arterial wall at the site of the angiomatoid lesions. Arterial wall destruction is a frequent finding in association with angiomatoid lesions. A dilatation lesion encircles the angiomatoid lesion












References
- ↑ Marcos E, Fadel E, Sanchez O, Humbert M, Dartevelle P, Simonneau G, Hamon M, Adnot S, Eddahibi S (May 2004). “Serotonin-induced smooth muscle hyperplasia in various forms of human pulmonary hypertension”. Circ. Res. 94 (9): 1263–70. doi:10.1161/01.RES.0000126847.27660.69. PMID 15059929.
- ↑ Eddahibi S, Fabre V, Boni C, Martres MP, Raffestin B, Hamon M, Adnot S (February 1999). “Induction of serotonin transporter by hypoxia in pulmonary vascular smooth muscle cells. Relationship with the mitogenic action of serotonin”. Circ. Res. 84 (3): 329–36. PMID 10024307.
- ↑ 3.0 3.1 3.2 3.3 Huber LC, Bye H, Brock M (2015). “The pathogenesis of pulmonary hypertension–an update”. Swiss Med Wkly. 145: w14202. doi:10.4414/smw.2015.14202. PMID 26479975.
- ↑ Peacock A (March 2013). “Pulmonary hypertension”. Eur Respir Rev. 22 (127): 20–5. doi:10.1183/09059180.00006912. PMID 23457160.
- ↑ Tuder RM, Stacher E, Robinson J, Kumar R, Graham BB (December 2013). “Pathology of pulmonary hypertension”. Clin. Chest Med. 34 (4): 639–50. doi:10.1016/j.ccm.2013.08.009. PMID 24267295.
- ↑ Michelakis ED (June 2014). “Pulmonary arterial hypertension: yesterday, today, tomorrow”. Circ. Res. 115 (1): 109–14. doi:10.1161/CIRCRESAHA.115.301132. PMID 24951761.
- ↑ Douwes JM, Berger RM (February 2011). “The maze of vasodilator response criteria”. Pediatr Cardiol. 32 (2): 245–6. doi:10.1007/s00246-010-9849-8. PMID 21110189.
- ↑ Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F (March 2015). “Endothelial-to-mesenchymal transition in pulmonary hypertension”. Circulation. 131 (11): 1006–18. doi:10.1161/CIRCULATIONAHA.114.008750. PMID 25593290.
- ↑ Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M (May 2016). “In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug”. Circulation. 133 (18): 1783–94. doi:10.1161/CIRCULATIONAHA.115.020617. PMC 4856565. PMID 27045138.
- ↑ Jobe AH, Bancalari E (June 2001). “Bronchopulmonary dysplasia”. Am. J. Respir. Crit. Care Med. 163 (7): 1723–9. doi:10.1164/ajrccm.163.7.2011060. PMID 11401896.
- ↑ Welsh CH, Hassell KL, Badesch DB, Kressin DC, Marlar RA (1996). “Coagulation and fibrinolytic profiles in patients with severe pulmonary hypertension”. Chest. 110 (3): 710–7. doi:10.1378/chest.110.3.710. PMID 8797416.
- ↑ Hoeper MM, Sosada M, Fabel H (1998). “Plasma coagulation profiles in patients with severe primary pulmonary hypertension”. Eur Respir J. 12 (6): 1446–9. doi:10.1183/09031936.98.12061446. PMID 9877507.
- ↑ Momose Y, Aimi Y, Hirayama T, Kataoka M, Ono M, Yoshino H; et al. (2015). “De novo mutations in the BMPR2 gene in patients with heritable pulmonary arterial hypertension”. Ann Hum Genet. 79 (2): 85–91. doi:10.1111/ahg.12096. PMID 25612240.
- ↑ Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G; et al. (2006). “BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension”. Hum Mutat. 27 (2): 212–3. doi:10.1002/humu.9398. PMID 16429403.
- ↑ Ghofrani HA, Seeger W, Grimminger F (2005). “Imatinib for the treatment of pulmonary arterial hypertension”. N Engl J Med. 353 (13): 1412–3. doi:10.1056/NEJMc051946. PMID 16192491.
- ↑ Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F; et al. (2001). “Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension”. J Clin Invest. 108 (8): 1141–50. doi:10.1172/JCI12805. PMC 209526. PMID 11602621.
- ↑ Rich, S.; Dantzker, DR.; Ayres, SM.; Bergofsky, EH.; Brundage, BH.; Detre, KM.; Fishman, AP.; Goldring, RM.; Groves, BM. (1987). “Primary pulmonary hypertension. A national prospective study”. Ann Intern Med. 107 (2): 216–23. PMID 3605900. Unknown parameter
|month=ignored (help) - ↑ ACCF/AHA 2009 Expert Consensus Document on Pulmonary Hypertension
- ↑ 19.0 19.1 Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM; et al. (2004). “Pathologic assessment of vasculopathies in pulmonary hypertension”. J Am Coll Cardiol. 43 (12 Suppl S): 25S–32S. doi:10.1016/j.jacc.2004.02.033. PMID 15194175.
Causes
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ralph Matar; Mohamed Moubarak, M.D. [2]; José Eduardo Riceto Loyola Junior, M.D.[3]
Overview
Pulmonary hypertension may be caused by either left heart failure (the most common cause) or other somewhat common causes such as HIV, systemic sclerosis, portal hypertension, congenital heart disease, and sickle cell disease. The World Health Organization (WHO) has classified PH based on etiology into five distinct groups: Group 1 (pulmonary arterial hypertension), Group 2 (PH due to left heart failure), Group 3 (PH due to chronic lung disease and/or hypoxemia), Group 4 (PH due to chronic thromboembolic disease), and Group 5 (PH due to multifactorial mechanisms).
Causes
Life Threatening Causes
Life-threatening causes include conditions which may result in death or permanent disability within 24 hours if left untreated.
- Pulmonary hypertension itself is not a life-threatening condition, but it is progressively fatal if left untreated. Pulmonary embolism and acute left heart failure are two causes of pulmonary hypertension that can be quickly fatal.
Common Causes
The most common cause of pulmonary hypertension is left heart failure leading to pulmonary venous hypertension. Other common causes of pulmonary arterial hypertension (PAH) include:[1][2][3][4]
- Cor pulmonale (right heart failure due to pulmonary disease)
- Congestive heart failure
- Congenital heart disease
- Chronic pulmonary thromboembolism
- COPD
- Familial Pulmonary Hypertension
- HIV
- Interstitial lung disease
- Mitral stenosis
- Obstructive sleep apnea
- Portal hypertension
- Pickwickian syndrome
- Right-sided valvular disease
- Systemic sclerosis
- Systemic lupus erythematosus
- Sickle cell disease
- Stimulant drugs such as amphetamines
Idiopathic Pulmonary Arterial Hypertension
When none of the causes on this page can be found, the disease is termed idiopathic pulmonary arterial hypertension (IPAH).
Causes by Organ System
Causes in Alphabetical Order
- Alveolar capillary dysplasia with misalignment of pulmonary veins
- Atrial Septal Defects
- Bronchiectasis
- Bronchopulmonary dysplasia
- Cholesterol ester storage disease
- Chronic hemolytic anemia
- Chronic obstructive pulmonary disease
- Chronic renal failure on dialysis
- Churg-Strauss syndrome
- Coal workers’ pneumoconiosis
- Cor triatriatum
- Cystic fibrosis
- Diastolic dysfunction
- Diethylpropion
- Fallot tetralogy
- Fetal circulation, persistent
- Fibrosing mediastinitis
- Gaucher disease
- Glycogen storage diseases
- High Altitude(chronically)
- Idiopathic pulmonary haemosiderosis
- Indian familial childhood cirrhosis
- Interstitial Lung Disease
- Idiopathic spinal scoliosis
- Langerhans cell histiocytosis
- Mitral valve insufficiency
- Mitral valve stenosis
- Monocrotaline poisoning
- Myeloproliferative disorders
- Neurofibromatosis
- Obstructive sleep apnea
- Paroxysmal nocturnal haemoglobinuria
- Pergolide
- Phentermine poisoning
- Pickwickian syndrome
- Portal hypertension
- Polycythemia vera
- Pulmonary alveolar microlithiasis
- Pulmonary capillary hemangiomatosis
- Pulmonary embolism
- Pulmonary fibrosis
- Pulmonary veno-occlusive disease
- Sarcoidosis
- Schistosoma japonicum
- Schistosoma mansoni
- Sickle cell disease
- Splenectomy
- Systemic lupus erythematosus
- Systolic dysfunction
- Tropical pulmonary eosinophilia
- Vasculitis
- Ventricular septal defect
Causes by Clinical Classification
Class 1: Pulmonary arterial hypertension
- Idiopathic pulmonary arterial hypertension
- Heritable (BMPR2, ALK-1, ENG, SMAD9, CAV1, KCNK3)
- Drug and toxin induced
Class 2: Pulmonary hypertension due to left heart disease
- Left ventricular systolic dysfunction
- Left ventricular diastolic dysfunction
- Valvular disease
- Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies
Class 3: Pulmonary hypertension due to lung diseases and/or hypoxia
- Chronic obstructive pulmonary disease
- Interstitial lung disease
- Other pulmonary diseases with mixed restrictive and obstructive pattern
- Sleep-disordered breathing
- Alveolar hypoventilation disorders
- Chronic exposure to high altitude
- Developmental lung diseases
Class 4: Chronic thromboembolic pulmonary hypertension
Class 5: Pulmonary hypertension with unclear multifactorial mechanisms
- Hematologic disorders: Chronic hemolytic anemia, Myeloproliferative disorders, splenectomy,
- Systemic disorders: Sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis
- Metabolic disorders: Glycogen storage disease, Gaucher disease, thyroid disorders
- Miscellaneous: Tumoral obstruction, fibrosing mediastinitis, chronic renal failure, segmental PH
References
- ↑ Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K; et al. (2004). “Pulmonary hypertension as a risk factor for death in patients with sickle cell disease”. N Engl J Med. 350 (9): 886–95. doi:10.1056/NEJMoa035477. PMID 14985486.
- ↑ Nayak NC, Chitale AR (2013). “Indian childhood cirrhosis (ICC) & ICC-like diseases: the changing scenario of facts versus notions”. Indian J Med Res. 137 (6): 1029–42. PMC 3734708. PMID 23852284.
- ↑ Schultze AE, Roth RA (1998). “Chronic pulmonary hypertension–the monocrotaline model and involvement of the hemostatic system”. J Toxicol Environ Health B Crit Rev. 1 (4): 271–346. doi:10.1080/10937409809524557. PMID 9776954.
- ↑ Kashyap S, Mohapatra PR (2013). “Pulmonary alveolar microlithiasis”. Lung India. 30 (2): 143–7. doi:10.4103/0970-2113.110424. PMC 3669555. PMID 23741096.
Differentiating Pulmonary hypertension from other Diseases
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Assistant Editor(s)-in-Chief: Ralph Matar, Rim Halaby
Overview
One of the most common initial presentations of patients with pulmonary hypertension is dyspnea; therefore, the differential diagnosis is very broad. As the disease progresses with time, more symptoms related to right ventricular hypertrophy and failure occur; which further narrows down the differential diagnosis.
Differential Diagnosis
Differentiating Pulmonary Arterial Hypertension from Other Diseases
Major entities in the differential diagnosis of pulmonary arterial hypertension are:[1]
- Chronic pulmonary thromboembolism
- Mitral stenosis
- Left atrial tumors
- Pulmonary veno-occlusive disease
Differentiating Shortness of Breath or Dyspnea from other Diseases
- The underlying causes of dyspnea are classified as acute causes and chronic causes based on the disease course. Different causes of dyspnea include pulmonary (upper and lower airway), cardiovascular, central nervous system, toxic and metabolic, and systemic diseases.
Diseases that cause shortness of breath have to be differentiated upon the following table[2]
To review the differential diagnosis of dyspnea and fever, click here.
To review the differential diagnosis of dyspnea and chest pain, click here.
To review the differential diagnosis of dyspnea and cough, click here.
To review the differential diagnosis of dyspnea and jugular vein distention, click here.
To review the differential diagnosis of dyspnea and cyanosis or clubbing, click here.
To review the differential diagnosis of dyspnea and loss of consciousness or agitation, click here.
To review the differential diagnosis of dyspnea with normal auscultation, click here.
To review the differential diagnosis of dyspnea with stridor, click here.
To review the differential diagnosis of dyspnea with wheezing, click here.
To review the differential diagnosis of dyspnea with crackle, click here.
To review the differential diagnosis of dyspnea with rhonchi, click here.
To review the differential diagnosis of dyspnea, fever, and cough, click here.
To review the differential diagnosis of dyspnea, fever, and chest pain, click here.
To review the differential diagnosis of dyspnea, cough, and cyanosis or clubbing click here.
To review the differential diagnosis of dyspnea, fever, chest pain, cough, and cyanosis or clubbing click here.
Abbreviations: ABG (arterial blood gas); ACE (angiotensin converting enzyme); BMI (body mass index); CBC (complete blood count); CSF (cerebrospinal fluid); CXR (chest X-ray); DOE (dyspnea on exercise); ECG (electrocardiogram); FEF (forced expiratory flow rate); FEV1 (forced expiratory volume); FVC (forced vital capacity); JVD (jugular vein distention); MCV (mean corpuscular volume); Plt (platelet); RV (residual volume); SIADH (syndrome of inappropriate antidiuretic hormone); TSH (thyroid stimulating hormone); Vt (tidal volume); WBC (white blood cell);
References
- ↑ Doi S (2008). “[Differential diagnosis of pulmonary hypertension]”. Nihon Rinsho. 66 (11): 2127–32. PMID 19051731.
- ↑ Berliner D, Schneider N, Welte T, Bauersachs J (2016). “The Differential Diagnosis of Dyspnea”. Dtsch Arztebl Int. 113 (49): 834–845. doi:10.3238/arztebl.2016.0834. PMC 5247680. PMID 28098068.
- ↑ Bernstein JA, Cremonesi P, Hoffmann TK, Hollingsworth J (2017). “Angioedema in the emergency department: a practical guide to differential diagnosis and management”. Int J Emerg Med. 10 (1): 15. doi:10.1186/s12245-017-0141-z. PMC 5389952. PMID 28405953.
- ↑ Bjornsson HM, Graffeo CS (2010). “Improving diagnostic accuracy of anaphylaxis in the acute care setting”. West J Emerg Med. 11 (5): 456–61. PMC 3027438. PMID 21293765.
- ↑ O’Horo JC, Rogus-Pulia N, Garcia-Arguello L, Robbins J, Safdar N (2015). “Bedside diagnosis of dysphagia: a systematic review”. J Hosp Med. 10 (4): 256–65. doi:10.1002/jhm.2313. PMC 4607509. PMID 25581840.
- ↑ Bjornson CL, Johnson DW (2013). “Croup in children”. CMAJ. 185 (15): 1317–23. doi:10.1503/cmaj.121645. PMC 3796596. PMID 23939212.
- ↑ Negus VE (1927). “The Function of the Epiglottis”. J Anat. 62 (Pt 1): 1–8. PMC 1250045. PMID 17104162.
- ↑ Meltzer EO, Hamilos DL (2011). “Rhinosinusitis diagnosis and management for the clinician: a synopsis of recent consensus guidelines”. Mayo Clin Proc. 86 (5): 427–43. doi:10.4065/mcp.2010.0392. PMC 3084646. PMID 21490181.
- ↑ 9.0 9.1 Wood RP, Milgrom H (September 1996). “Vocal cord dysfunction”. J. Allergy Clin. Immunol. 98 (3): 481–5. PMID 8828523.
- ↑ 10.0 10.1 Hodder R, Lougheed MD, Rowe BH, FitzGerald JM, Kaplan AG, McIvor RA (2010). “Management of acute asthma in adults in the emergency department: nonventilatory management”. CMAJ. 182 (2): E55–67. doi:10.1503/cmaj.080072. PMC 2817338. PMID 19858243.
- ↑ 11.0 11.1 Cantin, Luce; Bankier, Alexander A.; Eisenberg, Ronald L. (2009). “Bronchiectasis”. American Journal of Roentgenology. 193 (3): W158–W171. doi:10.2214/AJR.09.3053. ISSN 0361-803X.
- ↑ Molis MA, Molis WE (2010). “Exercise-induced bronchospasm”. Sports Health. 2 (4): 311–7. doi:10.1177/1941738110373735. PMC 3445098. PMID 23015953.
- ↑ 13.0 13.1 Holbro A, Lehmann T, Girsberger S, Stern M, Gambazzi F, Lardinois D, Heim D, Passweg JR, Tichelli A, Bubendorf L, Savic S, Hostettler K, Grendelmeier P, Halter JP, Tamm M (2013). “Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation”. Biol. Blood Marrow Transplant. 19 (6): 973–80. doi:10.1016/j.bbmt.2013.03.017. PMID 23562737.
- ↑ 14.0 14.1 Qureshi H, Sharafkhaneh A, Hanania NA (2014). “Chronic obstructive pulmonary disease exacerbations: latest evidence and clinical implications”. Ther Adv Chronic Dis. 5 (5): 212–27. doi:10.1177/2040622314532862. PMC 4131503. PMID 25177479.
- ↑ Dela Cruz CS, Tanoue LT, Matthay RA (2011). “Lung cancer: epidemiology, etiology, and prevention”. Clin Chest Med. 32 (4): 605–44. doi:10.1016/j.ccm.2011.09.001. PMC 3864624. PMID 22054876.
- ↑ Simonetti AF, Viasus D, Garcia-Vidal C, Carratalà J (2014). “Management of community-acquired pneumonia in older adults”. Ther Adv Infect Dis. 2 (1): 3–16. doi:10.1177/2049936113518041. PMC 4072047. PMID 25165554.
- ↑ Currie GP, Alluri R, Christie GL, Legge JS (2007). “Pneumothorax: an update”. Postgrad Med J. 83 (981): 461–5. doi:10.1136/pgmj.2007.056978. PMC 2600088. PMID 17621614.
- ↑ Bĕlohlávek J, Dytrych V, Linhart A (2013). “Pulmonary embolism, part I: Epidemiology, risk factors and risk stratification, pathophysiology, clinical presentation, diagnosis and nonthrombotic pulmonary embolism”. Exp Clin Cardiol. 18 (2): 129–38. PMC 3718593. PMID 23940438.
- ↑ Swart E, Laratta J, Slobogean G, Mehta S (February 2017). “Operative Treatment of Rib Fractures in Flail Chest Injuries: A Meta-analysis and Cost-Effectiveness Analysis”. J Orthop Trauma. 31 (2): 64–70. doi:10.1097/BOT.0000000000000750. PMID 27984449.
- ↑ 20.0 20.1 20.2 20.3 Bruyninckx R, Aertgeerts B, Bruyninckx P, Buntinx F (2008). “Signs and symptoms in diagnosing acute myocardial infarction and acute coronary syndrome: a diagnostic meta-analysis”. Br J Gen Pract. 58 (547): 105–11. doi:10.3399/bjgp08X277014. PMC 2233977. PMID 18307844.
- ↑ Gaggin, Hanna K.; Januzzi, James L. (2013). “Biomarkers and diagnostics in heart failure”. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. 1832 (12): 2442–2450. doi:10.1016/j.bbadis.2012.12.014. ISSN 0925-4439.
- ↑ 22.0 22.1 van Steijn JH, Sleijfer DT, van der Graaf WT, van der Sluis A, Nieboer P (2002). “How to diagnose cardiac tamponade”. Neth J Med. 60 (8): 334–8. PMID 12481882.
- ↑ Martindale, Jennifer L.; Noble, Vicki E.; Liteplo, Andrew (2013). “Diagnosing pulmonary edema”. European Journal of Emergency Medicine. 20 (5): 356–360. doi:10.1097/MEJ.0b013e32835c2b88. ISSN 0969-9546.
- ↑ Debiasi RL, Tyler KL (2004). “Molecular methods for diagnosis of viral encephalitis”. Clin Microbiol Rev. 17 (4): 903–25, table of contents. doi:10.1128/CMR.17.4.903-925.2004. PMC 523566. PMID 15489354.
- ↑ McAllister TW (2011). “Neurobiological consequences of traumatic brain injury”. Dialogues Clin Neurosci. 13 (3): 287–300. PMC 3182015. PMID 22033563.
- ↑ Peter JV, Sudarsan TI, Moran JL (2014). “Clinical features of organophosphate poisoning: A review of different classification systems and approaches”. Indian J Crit Care Med. 18 (11): 735–45. doi:10.4103/0972-5229.144017. PMC 4238091. PMID 25425841.
- ↑ Chin RL, Olson KR, Dempsey D (2007). “Salicylate toxicity from ingestion and continued dermal absorption”. Cal J Emerg Med. 8 (1): 23–5. PMC 2859737. PMID 20440389.
- ↑ Lane TR, Williamson WJ, Brostoff JM (2008). “Carbon monoxide poisoning in a patient with carbon dioxide retention: a therapeutic challenge”. Cases J. 1 (1): 102. doi:10.1186/1757-1626-1-102. PMC 2533003. PMID 18710551.
- ↑ Westerberg DP (March 2013). “Diabetic ketoacidosis: evaluation and treatment”. Am Fam Physician. 87 (5): 337–46. PMID 23547550.
- ↑ Taylor CB (2006). “Panic disorder”. BMJ. 332 (7547): 951–5. doi:10.1136/bmj.332.7547.951. PMC 1444835. PMID 16627512.
- ↑ Lee SY, Chien DK, Huang CH, Shih SC, Lee WC, Chang WH (August 2017). “Dyspnea in pregnancy”. Taiwan J Obstet Gynecol. 56 (4): 432–436. doi:10.1016/j.tjog.2017.04.035. PMID 28805596.
- ↑ Askim Å, Mehl A, Paulsen J, DeWan AT, Vestrheim DF, Åsvold BO; et al. (2016). “Epidemiology and outcome of sepsis in adult patients with Streptococcus pneumoniae infection in a Norwegian county 1993-2011: an observational study”. BMC Infect Dis. 16: 223. doi:10.1186/s12879-016-1553-8. PMC 4877975. PMID 27216810.
- ↑ Stang MT, Armstrong MJ, Ogilvie JB, Yip L, McCoy KL, Faber CN, Carty SE (July 2012). “Positional dyspnea and tracheal compression as indications for goiter resection”. Arch Surg. 147 (7): 621–6. doi:10.1001/archsurg.2012.96. PMID 22430090.
- ↑ Schwenk NR, Schapira RM, Byrd JC (November 1994). “Laryngeal carcinoma presenting as platypnea”. Chest. 106 (5): 1609–11. PMID 7956433.
- ↑ Conti V, Calia N, Pasquini C, Zardi S, Finetti C, Stomeo F, Ravenna F (April 2013). “[Chronic cough and worsening dyspnea: a case of idiopathic tracheal stenosis]”. Recenti Prog Med (in Italian). 104 (4): 156–8. doi:10.1701/1271.14026. PMID 23748638.
- ↑ Sharafkhaneh A, Hanania NA, Kim V (2008). “Pathogenesis of emphysema: from the bench to the bedside”. Proc Am Thorac Soc. 5 (4): 475–7. doi:10.1513/pats.200708-126ET. PMC 2645322. PMID 18453358.
- ↑ Sajkov D, Petrovsky N, Palange P (June 2010). “Management of dyspnea in advanced pulmonary arterial hypertension”. Curr Opin Support Palliat Care. 4 (2): 76–84. doi:10.1097/SPC.0b013e328338c1e0. PMID 20407377.
- ↑ Baughman RP, Shipley RT, Loudon RG, Lower EE (1991). “Crackles in interstitial lung disease. Comparison of sarcoidosis and fibrosing alveolitis”. Chest. 100 (1): 96–101. PMID 2060395.
- ↑ Moher D, Cole CW, Hill GB (November 1992). “Epidemiology of abdominal aortic aneurysm: the effect of differing definitions”. Eur J Vasc Surg. 6 (6): 647–50. PMID 1451823.
- ↑ Khanna D, Clements PJ, Furst DE, Chon Y, Elashoff R, Roth MD, Sterz MG, Chung J, FitzGerald JD, Seibold JR, Varga J, Theodore A, Wigley FM, Silver RM, Steen VD, Mayes MD, Connolly MK, Fessler BJ, Rothfield NF, Mubarak K, Molitor J, Tashkin DP (February 2005). “Correlation of the degree of dyspnea with health-related quality of life, functional abilities, and diffusing capacity for carbon monoxide in patients with systemic sclerosis and active alveolitis: results from the Scleroderma Lung Study”. Arthritis Rheum. 52 (2): 592–600. doi:10.1002/art.20787. PMID 15692967.
- ↑ Ziegler, Bruna; Rovedder, Paula Maria Eidt; Dalcin, Paulo de Tarso Roth; Menna-Barreto, Sérgio Saldanha (2009). “Padrões ventilatórios na espirometria em pacientes adolescentes e adultos com fibrose cística”. Jornal Brasileiro de Pneumologia. 35 (9): 854–859. doi:10.1590/S1806-37132009000900006. ISSN 1806-3713.
- ↑ Thomas R, Jenkins S, Eastwood PR, Lee YC, Singh B (July 2015). “Physiology of breathlessness associated with pleural effusions”. Curr Opin Pulm Med. 21 (4): 338–45. doi:10.1097/MCP.0000000000000174. PMC 5633324. PMID 25978627.
- ↑ Vodoz JF, Cottin V, Glérant JC, Derumeaux G, Khouatra C, Blanchet AS; et al. (2009). “Right-to-left shunt with hypoxemia in pulmonary hypertension”. BMC Cardiovasc Disord. 9: 15. doi:10.1186/1471-2261-9-15. PMC 2671488. PMID 19335916.
- ↑ Dubé BP, Dres M (2016). “Diaphragm Dysfunction: Diagnostic Approaches and Management Strategies”. J Clin Med. 5 (12). doi:10.3390/jcm5120113. PMC 5184786. PMID 27929389.
- ↑ Campbell IA, Bah-Sow O (2006). “Pulmonary tuberculosis: diagnosis and treatment”. BMJ. 332 (7551): 1194–7. doi:10.1136/bmj.332.7551.1194. PMC 1463969. PMID 16709993.
- ↑ Nakamura M, Satoh M, Kowada S, Satoh H, Tashiro A, Sato F, Masuda T, Hiramori K (February 2002). “Reversible restrictive cardiomyopathy due to light-chain deposition disease”. Mayo Clin. Proc. 77 (2): 193–6. doi:10.4065/77.2.193. PMID 11838655.
- ↑ Barstow C, McDivitt JD (March 2017). “Cardiovascular Disease Update: Bradyarrhythmias”. FP Essent. 454: 18–23. PMID 28266824.
- ↑ Jung HO (2012). “Pericardial effusion and pericardiocentesis: role of echocardiography”. Korean Circ J. 42 (11): 725–34. doi:10.4070/kcj.2012.42.11.725. PMC 3518705. PMID 23236323.
- ↑ Vodoz JF, Cottin V, Glérant JC, Derumeaux G, Khouatra C, Blanchet AS; et al. (2009). “Right-to-left shunt with hypoxemia in pulmonary hypertension”. BMC Cardiovasc Disord. 9: 15. doi:10.1186/1471-2261-9-15. PMC 2671488. PMID 19335916.
- ↑ Lechtzin N, Lange DJ, Davey C, Becker B, Mitsumoto H (January 2007). “Measures of dyspnea in patients with amyotrophic lateral sclerosis”. Muscle Nerve. 35 (1): 98–102. doi:10.1002/mus.20669. PMID 17029274.
- ↑ Schwarz MI, Matthay RA, Sahn SA, Stanford RE, Marmorstein BL, Scheinhorn DJ (January 1976). “Interstitial lung disease in polymyositis and dermatomyositis: analysis of six cases and review of the literature”. Medicine (Baltimore). 55 (1): 89–104. PMID 1246203.
- ↑ Heinicke K, Taivassalo T, Wyrick P, Wood H, Babb TG, Haller RG (2011). “Exertional dyspnea in mitochondrial myopathy: clinical features and physiological mechanisms”. Am J Physiol Regul Integr Comp Physiol. 301 (4): R873–84. doi:10.1152/ajpregu.00001.2011. PMC 3197343. PMID 21813873.
- ↑ Tarui S (1995). “Glycolytic defects in muscle: aspects of collaboration between basic science and clinical medicine”. Muscle Nerve Suppl. 3: S2–9. PMID 7603522.
- ↑ Lane R, Adams L (1993). “Metabolic acidosis and breathlessness during exercise and hypercapnia in man”. J Physiol. 461: 47–61. PMC 1175244. PMID 8350272.
- ↑ DePalo LR (May 2002). “Fatal dyspnea in a patient with renal failure”. Mt. Sinai J. Med. 69 (3): 113–20. PMID 12035070.
- ↑ Sengupta A, Saha K, Jash D, Banerjee SN (2013). “Dyspnea with anemia turned out to be a case of hereditary hemorrhagic telangiectasia”. Asian J Transfus Sci. 7 (1): 75–8. doi:10.4103/0973-6247.106745. PMC 3613670. PMID 23559772.
- ↑ Bailey PH (July 2004). “The dyspnea-anxiety-dyspnea cycle–COPD patients’ stories of breathlessness: “It’s scary /when you can’t breathe““. Qual Health Res. 14 (6): 760–78. doi:10.1177/1049732304265973. PMID 15200799.
- ↑ Perri GA (2013). “Ascites in patients with cirrhosis”. Can Fam Physician. 59 (12): 1297–9, e538–40. PMC 3860926. PMID 24336542.
- ↑ Neuman A, Gunnbjörnsdottir M, Tunsäter A, Nyström L, Franklin KA, Norrman E, Janson C (October 2006). “Dyspnea in relation to symptoms of anxiety and depression: A prospective population study”. Respir Med. 100 (10): 1843–9. doi:10.1016/j.rmed.2006.01.016. PMID 16516455.
- ↑ Qiabi M, Chagnon K, Beaupré A, Hercun J, Rakovich G (2015). “Scoliosis and bronchial obstruction”. Can Respir J. 22 (4): 206–8. PMC 4530852. PMID 26083538.
- ↑ Sin DD, Jones RL, Man SF (July 2002). “Obesity is a risk factor for dyspnea but not for airflow obstruction”. Arch. Intern. Med. 162 (13): 1477–81. PMID 12090884.
- ↑ Uyar M, Elbek O, Bakır K, Kibar Y, Bayram N, Dikensoy Ö (2012). “Churg-Strauss syndrome related to montelukast”. Tuberk Toraks. 60 (1): 56–8. PMID 22554368.
- ↑ Tilanus A, Van der Niepen P, Geers C, Wissing KM (2015). “Pulmonary Limited MPO-ANCA Microscopic Polyangiitis and Idiopathic Lung Fibrosis in a Patient with a Diagnosis of IgA Nephropathy”. Case Rep Nephrol. 2015: 378170. doi:10.1155/2015/378170. PMC 4525752. PMID 26266064.
- ↑ Cardenas-Garcia J, Farmakiotis D, Baldovino BP, Kim P (2012). “Wegener’s granulomatosis in a middle-aged woman presenting with dyspnea, rash, hemoptysis and recurrent eye complaints: a case report”. J Med Case Rep. 6: 335. doi:10.1186/1752-1947-6-335. PMC 3492078. PMID 23034218.
- ↑ Bal, Amanjit; Das, Ashim; Gupta, Dheeraj; Garg, Mandeep (2014). “Goodpasture’s Syndrome and p-ANCA Associated Vasculitis in a Patient of Silicosiderosis: An Unusual Association”. Case Reports in Pulmonology. 2014: 1–7. doi:10.1155/2014/398238. ISSN 2090-6846.
Epidemiology and Demographics
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ralph Matar; José Eduardo Riceto Loyola Junior, M.D.[2]
Overview
Pulmonary arterial hypertension has been considered as a disease of young women. The mean age of patients in the U.S. registry was 36 years and the overall female-to-male ratio was 1.7:1.
Epidemiology and Demographics
Incidence
- While previously considered a rare disease, the most recent evidence from a French registry suggests that the incidence of new cases of pulmonary arterial hypertension is 0.20-0.30 cases per 100,000 individuals.[1]
Prevalence
- The prevalence of pulmonary hypertension is approximately 1.5-5 per 100,000 individuals.[2]
Age
- Pulmonary hypertension usually develops between the ages of 20 and 60, but it can occur at any age. The mean age of patients in the U.S. registry was approximately 36 years old.[1]
Gender
- The female-to-male ratio for PH is approximately 1.7:1.[1]
- Idiopathic pulmonary hypertension (IPAH), which is more prevalent in women (3x more common), was considered the most common type of pulmonary arterial hypertension in a French registry. Usually it affects women between 30-60 years old.[2]
- Males often face a worse prognosis.[2]
References
- ↑ 1.0 1.1 1.2 Rich, S.; Dantzker, DR.; Ayres, SM.; Bergofsky, EH.; Brundage, BH.; Detre, KM.; Fishman, AP.; Goldring, RM.; Groves, BM. (1987). “Primary pulmonary hypertension. A national prospective study”. Ann Intern Med. 107 (2): 216–23. PMID 3605900. Unknown parameter
|month=ignored (help) - ↑ 2.0 2.1 2.2 Levine DJ (2021). “Pulmonary arterial hypertension: updates in epidemiology and evaluation of patients”. Am J Manag Care. 27 (3 Suppl): S35–S41. doi:10.37765/ajmc.2021.88609. PMID 33710842 Check
|pmid=value (help).
Risk Factors
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Assistant Editor(s)-in-Chief: Ralph Matar; Rim Halaby, M.D. [2]
Overview
Pulmonary hypertension (PH) is a multifactorial disease involving genetic and environmental risk factors. Risk factors for pulmonary arterial hypertension include BMPR2 mutation, connective tissue disease, HIV infection, portal hypertension, fenfluramine use, and congenital heart disease with a shunt. Left heart and lung diseases are risk factors for PH. Patients with a hypercoagulable state (such as the presence of lupus anticoagulant, deficiency of protein C, protein S, or antithrombin III, chronic inflammatory disorders, myeloproliferative syndromes, and splenectomy) are at an increased risk for chronic thromboembolic pulmonary hypertension.
Risk Factors
Risk factors of pulmonary hypertension are divided as follows:[1]
Pulmonary Arterial Hypertension
- BMPR2 gene mutation
- ALK1 gene mutation
- Connective tissue disease
- HIV infection
- Portal hypertension
- Congenital heart disease
- Schistosomiasis
- Chronic hemolytic anemia
- Drugs and toxins
Shown below is a table summarizing the list of drugs associated with PAH:
| Definite risk | Possible risk | Likely risk | Unlikely risk |
|
Pulmonary hypertension due to Left Heart Disease
Pulmonary Hypertension due to Lung Diseases and/or Hypoxia
- Alveolar hypoventilation disorders
- Chronic exposure to high altitude
- Chronic obstructive pulmonary disease
- Developmental abnormalities
- Interstitial lung disease
- Pulmonary diseases with mixed restrictive and obstructive pattern
- Sleep related breathing problems
Chronic Thromboembolic Pulmonary Hypertension
- Pulmonary embolism
- Hypercoagulability (lupus anticoagulant, deficiency of protein C, protein S, or antithrombin III, chronic inflammatory disorders, myeloproliferative syndromes, and splenectomy)
PH with Unclear and/or Multifactorial Mechanisms
- Myeloproliferative disease
- Splenectomy
- Sarcoidosis
- Histiocytosis
- Lymphangioleiomyomatosis
- Neurofibromatosis
- Vasculitis
- Glycogen storage disease
- Gaucher disease
- Thyroid disorder
- Obstruction by tumor
- Fibrosing mediastinitis
- Chronic renal failure on dialysis
References
- ↑ McLaughlin, VV.; Archer, SL.; Badesch, DB.; Barst, RJ.; Farber, HW.; Lindner, JR.; Mathier, MA.; McGoon, MD.; Park, MH. (2009). “ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association”. Circulation. 119 (16): 2250–94. doi:10.1161/CIRCULATIONAHA.109.192230. PMID 19332472. Unknown parameter
|month=ignored (help)
Screening
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Assistant Editor(s)-in-Chief: José Eduardo Riceto Loyola Junior, M.D.[2]; Ralph Matar
Overview
Patients with a known BMPR2 mutation, scleroderma, and portal hypertension undergoing evaluation for liver transplantation should receive periodic screening for pulmonary hypertension (PH) through echocardiography.
Screening
- Studies have not shown an impact on outcomes with pulmonary hypertension screening;
- Despite that, there is an expert consensus that some groups of patients must be screened for pulmonary hypertension such as:
- Patients with scleroderma spectrum disorders (especially the ones with corrected DLCO less than 80%);
- Patients with mutations for a heritable form of PAH;
- Patients with portal hypertension being considered for organ transplantation;
- These patients must be screened annually with echocardiography.[1]
Summary
Shown below is a table summarizing the recommended screening in several medical conditions associated with elevated risk for PH.[2][3]
| Condition | Recommended screening |
| Known BMPR2 mutation | Echocardiogram (yearly) |
| BMPR2 mutation in a first degree relative | Genetic counseling BMPR2 genotyping |
| Family history for PAH in 2 or more relatives | Genetic counseling BMPR2 genotyping |
| Systemic sclerosis | Echocardiogram (yearly) |
| Portal hypertension | Echocardiogram if orthotopic liver transplantation is in consideration |
| Sickle cell disease | Echocardiogram (yearly) |
| Previous use of fenfluramine | Echocardiogram in case of symptoms |
| Congenital heart disease | Echocardiogram at the time of diagnosis |
Echocardiography findings
Echocardiography findings that are suggestive of PH include:[4]
- Enlargement of the size of right atrium and right ventricle
- Decrease in the function of the right ventricle
- Displacement of the interventricular septum
- Tricuspid regurgitation
- Presence of pericardial effusion
References
- ↑ Poch D, Mandel J (2021). “Pulmonary Hypertension”. Ann Intern Med. 174 (4): ITC49–ITC64. doi:10.7326/AITC202104200. PMID 33844574 Check
|pmid=value (help). - ↑ ACCF/AHA 2009 Expert Consensus Document on Pulmonary Hypertension
- ↑ ACCF/AHA 2009 Expert Consensus Document on Pulmonary Hypertension
- ↑ ACCF/AHA 2009 Expert Consensus Document on Pulmonary Hypertension
Natural History, Complications and Prognosis
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Assistant Editor(s)-in-Chief: Ralph Matar; José Eduardo Riceto Loyola Junior, M.D.[2]
Overview
Pulmonary hypertension most common initial symptoms are dyspnea, fatigue, and syncope. There was an estimated median survival of 2.8 years for symptomatic patients who do not receive any treatment, with the most common cause of death as cor pulmonale, but survival rates have been increasing as new forms of treatment become available. Despite such advances, prognosis is still poor.
Natural History
- The National Institutes of Health (NIH) Registry estimated a median survival of 2.8 years for symptomatic patients with idiopathic pulmonary hypertension who do not receive any treatment, with the cause of death usually being right ventricular failure (cor pulmonale).[1]
- The 1, 3,and 5-year survival rates for untreated patients with idiopathic pulmonary hypertension were 68%, 48%, and 34%, respectively.[1]
- The median survival duration was even lower for patients with pulmonary hypertension that was associated with other diseases like portal hypertension, and scleroderma (2-year survival of 40% if untreated).
- A recent outcome study of those patients who had started treatment with bosentan demonstrated that 86% patients were alive at 3 years.
- With multiple agents now available, combination therapy is increasingly used. Impact of these agents on survival is not known, since many of them have been developed only recently. It would not be unreasonable to expect median survival to extend past 10 years in the near future.
Complications
Complications that can develop as a result of pulmonary hypertension are:[2]
- Right-sided heart failure (cor pulmonale)
- Blood clots
- Arrhythmia
- Hemoptysis
Prognosis
The prognosis of pulmonary hypertension is poor, but good with treatment. Without treatment, pulmonary hypertension will result in cor pulmonale. Pulmonary hypertension is associated with a 7 year mortality of 49%.
- The long-term prognosis has been known to be poor, however outcomes have changed dramatically over the last two decades. This may be attributed to the use of newer drug therapy, better overall care, and earlier diagnosis (lead time bias).
- Elevated pulmonary arterial pressure on heart catheterization is a predictor of mortality, especially if it is happening not only in the setting of myocarditis or decreased right ventricular ejection fraction, but also COPD. Treatment of pulmonary hypertension in these morbidities does not affect outcomes.[3]
- Survival rate at 5 years is 57%.[3]
- Persistently elevated BNP or NT-proBNP levels, PAH associated with connective tissue disease or portal hypertension indicate poorer prognosis.[3]
- Some people with this condition may have heart failure that could lead to death. Assessment of prognosis in patients with pulmonary arterial hypertension (PAH) is important since it influences both medical therapy and referral for transplantation.
- Mortality rate is 5.2-5.4 per 100,000 and is more common in African-Americans and women.
- The most common cause of hospitalization is heart failure.
- The most common cause of death is right ventricular failure and not chronic lower respiratory disease as was once thought.
- Surprisingly, patients with Eisenmenger syndrome have a more favorable hemodynamic profile and prognosis than adults with idiopathic or primary pulmonary hypertension.[4]
Indicators of Poor Prognosis
Indicators of poor prognosis include:
- Poor functional class[2]
- Poor exercise tolerance as assessed by the 6-minute-walk distance (6MWD)[5]
- Elevated brain natriuretic peptide (BNP)[6] or NT-proBNP levels.[3]
- Pericardial effusion[6]
- Persistently elevated right atrial size and pressure[7]
- Significant right ventricular dysfunction or failure[6]
- Low cardiac index[7]
- Underlying connective tissue disease[6]
- Decreased predicted carbon monoxide diffusing capacity[6]
- PAH associated with connective tissue disease or portal hypertension.
References
- ↑ 1.0 1.1 Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM; et al. (1987). “Primary pulmonary hypertension. A national prospective study”. Ann Intern Med. 107 (2): 216–23. PMID 3605900.
- ↑ 2.0 2.1 McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T; et al. (2004). “Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines”. Chest. 126 (1 Suppl): 78S–92S. doi:10.1378/chest.126.1_suppl.78S. PMID 15249497.
- ↑ 3.0 3.1 3.2 3.3 Poch D, Mandel J (2021). “Pulmonary Hypertension”. Ann Intern Med. 174 (4): ITC49–ITC64. doi:10.7326/AITC202104200. PMID 33844574 Check
|pmid=value (help). - ↑ Hopkins WE,Ochoa LL, Richardson GW, Trulock EP(1996) Comparison of the hemodynamics and survival or patients with severe pulmonary hypertension or Eisenmenger Syndrome.
- ↑ Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB; et al. (1996). “A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension”. N Engl J Med. 334 (5): 296–301. doi:10.1056/NEJM199602013340504. PMID 8532025.
- ↑ 6.0 6.1 6.2 6.3 6.4 Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS; et al. (2010). “Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL)”. Circulation. 122 (2): 164–72. doi:10.1161/CIRCULATIONAHA.109.898122. PMID 20585012.
- ↑ 7.0 7.1 D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM; et al. (1991). “Survival in patients with primary pulmonary hypertension. Results from a national prospective registry”. Ann Intern Med. 115 (5): 343–9. PMID 1863023.
Diagnosis
Diagnosis
Diagnostic Study of Choice | History & Symptoms | Physical Examination | Laboratory Findings | Electrocardiography | Chest x-ray | CT | MRI | Echocardiography or Ultrasound | Right heart catheterization | Other Diagnostic Studies
Treatment
Treatment
Medical Therapy | Surgery | Primary Prevention | Secondary Prevention | Cost-Effectiveness of Therapy | Future or Investigational Therapies
Looking for the patient version?
© 2026 MyEClinic – IFTM Institut für Telematik in der Medizin GmbH